
AUTORES:
Estudiantes de la especialidad de Fisioterapia y Rehabilitación del Instituto de la Clínica Ricardo Palma de Lima - Perú
Definición
Es una enfermedad de las células nerviosas motoras en el cerebro y la médula espinal que ocasiona la pérdida progresiva del control motor.
Se conocen tres tipos de ELA:
- Esporádica: es la forma más frecuente, representando aproximadamente el 90% de los casos. La enfermedad se produce de manera aleatoria (por azar), sin ninguna causa conocida y sin que haya otras personas de la familia afectada.
- Familiar: la enfermedad parece que tiene un componente hereditario.
- Territorial o Guameña: por observarse una frecuencia de la enfermedad (incidencia) extremadamente elevada en Guam (Pacífico).
El ELA, o Esclerosis Lateral Amiotrófica, es una enfermedad de las partes del sistema nervioso que controlan el movimiento de los músculos voluntarios.
La palabra amiotrófica significa "sin nutrimento muscular" y se refiere a la pérdida de las señales que los nervios envían normalmente a los músculos. Lateral significa "al lado" y se refiere a la ubicación del daño en la médula espinal. Esclerosis significa "endurecimiento" y se refiere al estado endurecido de la médula espinal en el Ela avanzada.
En Estados Unidos, el Ela se conoce también como enfermedad de Lou Gehrig, en honor del jugador de béisbol de los Yanquis que murió en 1941 debido a ese mal. En Inglaterra y en otras partes del mundo, se le conoce frecuentemente como enfermedad de las neuronas motoras, refiriéndose a las células que se pierden en esta enfermedad.
Las células que controlan los músculos, o neuronas motoras, se dividen en dos clases. Las neuronas motoras superiores se localizan en la parte superior del cerebro y ejercen algún control sobre las neuronas motoras inferiores ubicadas en el tronco cerebral y la médula espinal.
Las neuronas motoras inferiores están unidas directamente a los músculos mediante "cables” denominados axones. Haces de estos axones salen de la médula espinal y se extienden hacia los musculos. Son estos haces a los que se refieren los médicos cuando hablan de los "nervios".
La función de las neuronas motoras inferiores es directa. Envían señales de "funcionar" a los músculos. Cuando estas células mueren gradualmente, como sucede en el Ela, los músculos se debilitan progresivamente cada vez más y eventualmente, no pueden moverse (se paralizan).
Las neuronas motoras inferiores que controlan la mayor parte del cuerpo se encuentran en la médula espinal. Las que controlan los músculos involucrados al hablar, deglutir y en las expresiones faciales se encuentran en el tronco cerebral. Algunas veces se denominan neuronas motoras bulbares, debido a que la parte del tronco cerebral que las contiene tiene forma parecida a un bulbo. El término involucración bulbar significa que la enfermedad afecta a los músculos de la cara, boca y garganta.
Las neuronas motoras superiores tienen funciones más complejas. Es más difícil estudiarlas y no se les conoce suficientemente, aunque esto está cambiando con las técnicas nuevas.
Estas células parecen ejercer un control complejo sobre las neuronas motoras inferiores, permitiendo que los movimientos musculares sean uniformes, dirigidos y con intensidad diversa. (Por ejemplo, forman parte de un sistema elaborado que permite a la persona dirigir la mano a un vaso de agua, recogerlo, calcular su peso, utilizar la cantidad adecuada de fuerza para su peso y levantarlo hacia su boca, todo al mismo tiempo en que piensa otra cosa). Cuando se pierden las neuronas motoras superiores y quedan las neuronas motoras inferiores, todavía pueden hacerse movimientos, pero pueden volverse "tensos”.
Por lo general, en el Ela se observa una combinación de estos efectos debido a que tanto las neuronas superiores como las inferiores están muriendo. Las personas con ELA pueden tener músculos débiles y degenerados, acompañados por tensión (espasticidad). Las sacudidas y calambres musculares son comunes; ocurren porque los axones degenerados (nervios) se vuelven "irritables".
ETIOLOGIA
La esclerosis lateral amiotrófica es causada por la pérdida progresiva de los nervios motores en la médula espinal y el cerebro. En aproximadamente el 10% de los casos es causada por un defecto genético, mientras que en otros casos se desconoce la causa del deterioro del nervio.
La esclerosis lateral amiotrófica afecta aproximadamente a 1 de cada 100.000 personas y, aparte de los antecedentes familiares, no se conocen otros factores de riesgo.
La ELA aparece generalmente a finales de la edad mediana (un promedio es a finales de la quinta década de vida) o posteriormente, aunque ha habido casos de ELA en adultos jóvenes e inclusive en niños, así como también en personas de edad muy avanzada. Algunas formas genéticas de ELA tienen su inicio en la juventud.
Los hombres son un poco más susceptibles a tener ELA que las mujeres. Algunos estudios sugieren una proporción general de 1.2 hombres aproximadamente por cada mujer que tiene la enfermedad.
Los factores genéticos forman parte del cuadro de la ELA y la enfermedad puede presentarse en familias.
Con respecto a los datos estadísticos más importantes podemos destacar:
- El 50% de los pacientes mueren en el plazo de 18 meses del diagnóstico.
- El 80% mueren en el plazo de 5 años del diagnóstico.
- 10% viven más de 10 años.
- Se da más en hombres (tres hombres por cada dos mujeres).
- Pasados los sesenta años, la proporción hombre mujer es la misma.
- La edad media de inicio son los 55 años.
- El 80% de los casos comienzan entre las edades de 40 a 70 años.
- Los casos diagnosticados entre los 20 y los 40 años tienen una posibilidad más alta de sobrevivir más de 5 años.
Durante años los expertos han tratado de encontrar factores comunes a las personas que desarrollan ELA, tales como toxinas ambientales, peligros relacionados con ocupaciones, lugares de trabajo o residencia y así sucesivamente. Hasta ahora, la evidencia de dichos factores de riesgo y detonantes ha sido frustrantemente poco clara, aunque un descubrimiento reciente de una asociación entre desarrollar ELAy haber prestado servicio militar en la Guerra del Golfo a principios de los años noventa, ha indicado uno de los más fuertes de estos factores propuestos de riesgo.
Hace años, se creía generalmente que podría haber una sola causa que explicara todos los casos de ELA. Hoy en día, los médicos y científicos saben que éste no puede ser el caso. Juntos, trabajan para identificar las causas múltiples de la enfermedad.
Desde principios de los años noventa, han surgido diversos indicios para solucionar el problema de la causalidad de la ELA, y la mayoría de los expertos considera que estos indicios están relacionados entre sí. Especialistas en ELA estudian las causas probables siguientes:
Radicales Libres
Los radicales libres son moléculas que portan cargas eléctricas que los hacen inestables y responsables de daño a las estructuras celulares. Son una parte normal de la vida celular y las células pueden neutralizar generalmente a la mayoría de ellos y mantener su numero bajo control. Pero en la ELA, los radicales libres se acumulan hasta alcanzar niveles tóxicos y dañan las células, mediante un proceso de ataque denominado estrés oxidante.
Exceso de Glutamato
El glutamato es una sustancia química común en el sistema nervioso que las neuronas utilizan para enviar señales a otras neuronas. Pero, como muchas cosas, el glutamato tiene que estar presente en la cantidad correcta para funcionar: demasiado poco induce una carencia de señales, y una cantidad excesiva induce la muerte de las células nerviosas que reciben la señal.
La evidencia que proporcionan estudios de personas con ELA apunta hacia una sobreabundancia de glutamato en el sistema nervioso. Esto puede deberse al transporte inadecuado de glutamato lejos de las células nerviosas después de que ha terminado su función de envío de señales.
Los experimentos sugieren que un defecto podría deberse también al exceso de producción o liberación de glutamato por las células transmisoras o que podría deberse a defectos en los receptores de glutamato en las células receptoras.
Formación de Neurofilamentos
Las proteínas conocidas como neurofilamentos construyen el "andamiaje" que ayuda a las células nerviosas a mantener su forma. En las neuronas motoras afectadas por la ELA, estos neurofilamentos tienden a amontonarse cerca del cuerpo de la célula en vez de bajar por la "cola" (axón) de la célula, lo que podría ocasionar un "embotellamiento" en el tráfico celular.
Todavía no se determina la relación del depósito de neurofilamentos con la causalidad de ELA o el curso de la enfermedad.
Defectos en las Mitocondrias
De todas las partes funcionales de una célula, las "fábricas" que producen energía conocidas como mitocondrias son discutiblemente las más decisivas — especialmente para las células de alta energía como las neuronas motoras.
Se encuentran también entre las partes más complejas, menos conocidas y más estudiadas de la célula.
Las mitocondrias tienen su propio material genético (ADN). Se parece un poco al otro ADN de la célula que se organiza en cromosomas en el núcleo de la célula. Pero el ADN mitocondriaco se organiza de manera diferente, formando paquetes de anillos microscópicos de material genético que carece de muchas de las protecciones contra daños que poseen los cromosomas del núcleo.
Por ese motivo, y debido a que los procesos que se realizan en el interior de las mitocondrias producen radicales libres peligrosas, el ADN mitocondriaco está siempre en peligro de sufrir daños. Una cantidad determinada de daño ocurre como parte del proceso de envejecimiento, pero en la ALS, puede haber más daño a las mitocondrias que el daño promedio que sufren las células que envejecen. Todavía debe investigarse minuciosamente si este daño es el resultado o una causa — o ambos — de la enfermedad.
Suicidio Celular
La mayoría de las células poseen un programa integrado de "suicidio", conocido como muerte celular programada o apoptosis. En ciertas circunstancias, la muerte celular programada es normal. Pero en la ELA y otras enfermedades degenerativas, es posible que el programa de muerte celular se active inadecuadamente.
Anomalías del Sistema Inmune
Muchos trastornos que afectan el sistema nervioso son autoinmunes por naturaleza, lo que significa que se presentan cuando el sistema inmune del cuerpo ataca por equivocación, a sus propios tejidos. La autoinmunidad podría desempeñar un papel en la ELA, pero hasta ahora, faltan estudios concluyentes y ninguno de los tratamientos utilizados para otras enfermedades autoinmunes ha sido eficaz contra la ELA
Virus y Otros Agentes Infecciosos
Durante décadas, los científicos han supuesto que los virus pueden desempeñar un papel en la ELA y otras enfermedades que implican la degeneración de las células nerviosas. Parece lógico, pero hasta ahora, no hay pruebas de un detonante viral.
El virus de VIH (virus de inmunodeficiencia humana) que causa el SIDA (síndrome de inmunodeficiencia adquirida) puede ocasionar un síndrome parecido a la ELA que mejora mediante tratamiento con fármacos antivirales. El VIH es una causa infrecuente de ELA (la mayoría de los pacientes de ELA no son VIH-positivos), pero la conexión apoya la idea de que otros virus podrían también causar daño a las neuronas motoras.
En un estudio de ELA, se encontraron rastros de un tipo de virus denominado ecovirus en tejido de médula espinal, pero los estudios subsiguientes no han podido confirmar hasta ahora este descubrimiento. Años de estudio no han producido prueba de ninguna conexión entre la exposición al virus de la poliomielitis y el desarrollo de ELA, pero los virus siguen en la lista de detonantes potenciales de la ELA
Podrían implicarse otros microorganismos en ELA. La enfermedad de Lyme, un trastorno bacteriano transmitido por garrapatas infectadas, puede dañar las neuronas motoras.
Los priones, proteínas que pueden actuar como virus, cambian la forma de otras proteínas, convirtiéndolas de moléculas benéficas en moléculas altamente tóxicas. Los priones parecen preferir el sistema nervioso, por lo que se les examina en cuanto a un probable papel en ELA.
Toxinas
Los metales pesados, plomo, mercurio y arsénico, aunque pueden ser tóxicos para el sistema nervioso, rara vez, si acaso, son agentes causales en la ELA
El plomo puede dañar las neuronas superiores e inferiores, pero en Estados Unidos se ha monitoreado y restringido la exposición al plomo en la mayoría de las personas durante 25 años aproximadamente. En algunas circunstancias, puede valer la pena realizar estudios respecto a estas exposiciones.
El contacto prolongado con químicos agrícolas, tales como pesticidas, puede ser un detonante de ELA en algunos casos.
Puede producir algunos indicios la asociación del servicio en la Guerra del Golfo con la ALS, así como un grupo de casos de ELA encontrados en Texas.
Una alta incidencia de ELA en la Isla de Guam condujo a la idea de que la semilla cicadácea, ingerida en la isla, podría ser un detonante de ELA. Existen pruebas recientes que sugieren que el detonante real puede ser la ingestión de murciélagos que comen las semillas cicadáceas y que permiten probablemente que se acumulen hasta niveles tóxicos en su carne. La incidencia de ELA ha disminuido en la medida en que se ha reducido el consumo de murciélagos.
Lesiones Causadas por Descarga Eléctrica
Algunas personas han desarrollado ELA después de sufrir lesiones causadas por descarga eléctrica. Se carece de estudios referentes a estos casos.
Genes
Además de los genes que pueden inducir directamente la ELA, existen casi con certeza factores genéticos de riesgo o "factores de susceptibilidad" que pueden influir en que alguna persona desarrolle ELA en presencia de una segunda o tercera circunstancia (por ejemplo, la exposición a cierto virus o sustancia ambiental).
¿Es Hereditario?
La ELA es "familiar" — es decir, muestra un historial familiar — aproximadamente en un 10 por ciento de los casos. Se han identificado diversos genes relacionados con ALS o por lo menos, se los ha mapeado hacia una zona específica de un cromosoma.
El Gene SOD1
En 1993, investigadores apoya
Las mutaciones en este gene SOD1 son la causa de 10 a 20 por ciento de los casos familiares de ELA y tal vez también de 1 a 3 por ciento de los casos sin antecedentes familiares. (Ya que la ELA puede ser una enfermedad con un inicio muy tardío, algunas personas con mutaciones del SOD1 mueren probablemente por otras causas sin haber desarrollado nunca ELA, de manera que la falta de antecedentes familiares de ELA puede ser engañosa.
Las mutaciones del SOD1 conllevan generalmente a ELA heredado en un patrón autosomal dominante, lo que significa que la imperfección no reside en un cromosoma sexual (reside en un "autosoma") y que se requiere una imperfección en solamente uno de los dos genes SOD1 de una persona para ocasionar la enfermedad.
A veces, la ELA relacionada con el SOD1 sigue un patrón hereditario autosomal recesivo, lo que significa que se requieren dos genes con mutación — uno de cada padre — antes de que aparezcan los síntomas.
Otros Genes que Ocasionan la ALS
Se han observado otros genes que, al experimentar imperfecciones en los cromosomas 2, 9, 15, 18 y el cromosoma X, pueden conllevar a la ELASe espera obtener más mapeos, identificaciones y más conocimientos respecto a genes específicos, a medida en que se prosiga con las investigaciones.
La mayoría de las mutaciones X-ligadas afecta a varones (quienes cuentan con sólo un cromosoma X, apareado con un cromosoma Y), pero dichas mutaciones rara vez afectan a mujeres (quienes tienen dos cromosomas X, uno de los cuales generalmente no porta la mutación). Sin embargo, en ocasiones las mutaciones X-ligadas sí pueden afectar a mujeres.
Algunas mutaciones conllevan a las formas de ELA que se manifiestan en jóvenes, es decir, las que inician en la niñez o adolescencia. Estos trastornos son muy poco comunes.
Estudios Genéticos y Asesoría
La mayoría de los laboratorios comerciales realizan solamente estudios para detectar las mutaciones de los genes SOD1. Puede disponerse de otros tipos de pruebas mediante estudios especiales, para personas con síntomas o para las que tengan antecedentes familiares de ELA que se encuentren en la etapa de toma de decisiones respecto a la planeación familiar.
Las mutaciones del SOD1 pueden heredarse, por supuesto, de padres a hijos.
En la ELA deben considerarse cuidadosamente las implicaciones de los estudios genéticos. Es muy recomendable discutir estas repercusiones con el equipo de su clínica de la MDA o el centro de MDA/ALS, especialmente con su asesor genético. Dichas discusiones deben efectuarse antes de que se lleven a cabo los estudios y otra vez, cuando se reciban los resultados.
Diagnóstico, signos y exámenes
En muchas ocasiones, se llega al diagnóstico de Esclerosis Lateral Amiotrófica después de un tiempo prolongado y por un proceso de exclusión. Los síntomas iniciales de la enfermedad son muy parecidos a los que se presentan en otras enfermedades de origen neuromuscular.
Para poder realizar un diagnóstico de ELA es necesario:
Presencia de
- Degeneración de la motoneurona inferior (MNI), según criterios clínicos y electrofisiológicos.
- Degeneración de la motoneurona superior (MNS) según examen neurológico.
- Diseminación progresiva de los síntomas o signos en la región afectada o la progresión hacia otras regiones.
Ausencia de
- Evidencia de otro proceso que pudiera explicar los signos de degeneración de la motoneurona inferior o superior.
- Evidencia a través de neuroimágenes de otro proceso que pudiera explicar los signos clínicos y electrofisiológicos observados.
Los signos clínicos de degeneración o afectación de la motoneurona inferior (MNI) son:
- Debilidad muscular.
- Atrofia muscular.
- Fasciculaciones.
Los signos clínicos de degeneración o afectación de la motoneurona superior (MNS) son:
- Hiperreflexia: exageración de los reflejos.
- Contracciones rítmicas e involuntarias en un músculo o grupo muscular por la extensión brusca y pasiva de sus tendones (Clonus).
- Espasticidad.
- Pérdida de los reflejos cutáneo – abdominales.
Una serie de signos que verificará el médico en la exploración: signo de Babinski (Flexión dorsal de los dedos del pie al estimular la planta), signo de Hoffmann (la presión sobre la uña del dedo índice de la mano produce de forma refleja flexión de los otros dedos), etc.
El único examen complementario con valor equivalente a la clínica en el diagnóstico de la ELA es el electromiograma (registro gráfico de las corrientes eléctricas producidas por la contracción muscular o de la reacción de un músculo al estímulo eléctrico).
Niveles de certeza diagnóstica:
Según la combinación de signos de MNS y MNI se definen los siguientes niveles de certeza.
- ELA Clínicamente Definida
Evidencia clínica (signos) de MNS y MNI en tres regiones. Se consideran cuatro regiones:
- Bulbar.
- Cervical.
- Torácica
- Lumbar
- ELA Clínicamente Probable
Evidencia clínica (signos) de MNS y MNI en al menos dos regiones con algunos signos de MNS.
- ELA Clínicamente Probable – Apoyada por Laboratorio
Cuando los signos clínicos de MNS y MNI se encuentran en una sola región o cuando los signos de MNS están presentes en una región y los signos de MNI se hallan presentes en al menos dos miembros y otras causas han sido excluidas con estudios de laboratorio y neuroimágenes.
- ELA Clínicamente Posible
Cuando los signos clínicos de MNS y MNI se dan en una sola región o los signos de MNS se manifiestan en dos o más regiones. Otros diagnósticos deben excluirse para que el de "ELA Clínicamente Posible" sea aceptado.
- ELA Clínicamente Sospechosa
Se trata de un síndrome puro de MNI en el que las características clínicas no permiten considerar la posibilidad de ELA para que el paciente sea incluido en un ensayo terapéutico de la enfermedad.
Antes de comunicar el diagnóstico, debe ser confirmado por el neurólogo y éste tiene que tener en cuenta que el comunicar la enfermedad forma parte del cuidado de ella. El médico deberá tener en cuenta las características individuales del paciente y facilitar la información como un proceso. Toda persona adulta tiene derecho a conocer su diagnóstico y tomar decisiones sobre su terapéutica futura. Hay que tener en cuenta que la forma en que se comunique la enfermedad puede afectar el estado emocional del paciente y la forma en que encare el futuro. Parece conveniente poner nombre a la enfermedad y su característica de progresiva, pero remarcando aspectos positivos y la gran variabilidad individual, explicar la terapéutica disponible, sus efectos adversos y su efectividad real, los tratamientos en investigación y las terapias alternativas. Hay que dar información sobre las asociaciones de pacientes existentes y la posibilidad de una segunda opinión.
Según un estudio, hay tres objetivos que deben seguirse en el proceso de información del diagnóstico:
- No negarle información si la requiere.
- No imponer al paciente información que no desea escuchar.
- Evaluar y responder a las reacciones del paciente ante la información suministrada.
En resumen:
El examen neuromuscular indica debilidad, que a menudo empieza en un miembro o en grupos proximales (hombros, caderas). Puede haber temblores musculares, espasmos, fasciculación (contracciones) o atrofia muscular (pérdida de tejido). Es común que se presente atrofia y torcimiento de la lengua.
Asimismo, se puede presentar marcha rígida o torpe. Los reflejos pueden ser anormales, incluyendo la pérdida del reflejo nauseoso. Algunos pacientes presentan "incontinencia emocional" en la cual es difícil controlar el llanto o la risa.
- Una electromiografía revela que los nervios motores no están funcionado, aunque los nervios sensoriales estén normales.
- Una TC o una IRM se puede hacer para descartar otras condiciones.
- En caso de antecedentes familiares, se pueden hacer pruebas genéticas.
- Se puede hacer un examen de la respiración para verificar si los músculos respiratorios está afectados.
- Los exámenes de sangre pueden excluir otras condiciones que pueden causar síntomas similares.
MODO DE APARICIÓN
En la ELA se pierden gradualmente las células nerviosas que controlan las células musculares. En la mayoría de los casos, se desconoce la causa. A medida que se pierden estas neuronas motoras, los músculos que controlan se debilitan y después, dejan de funcionar. Eventualmente, la persona con ELA se paraliza.
La muerte, ocasionada generalmente por complicaciones respiratorias, sobreviene entre tres y cinco años después del diagnóstico (en algunos estudios se dice que después de que se observan los síntomas, de manera que no se sabe con certeza el tiempo). Aproximadamente el 10 por ciento de las personas con la enfermedad viven más de 10 años y algunas sobreviven durante décadas.
Es importante observar que la ELA no afecta directamente los músculos involuntarios, tales como aquéllos que controlan los latidos del corazón, el tracto gastrointestinal, la función intestinal, las funciones sexuales y de la vejiga. (Sin embargo, la incapacidad prolongada de movimientos y otros efectos de la ALS pueden tener algún impacto indirecto).
El dolor no es un componente principal del trastorno, aunque puede presentarse un dolor moderado como resultado de la inmovilidad y sus complicaciones diversas.
El oído, la vista, el tacto y la capacidad intelectual siguen siendo generalmente bastante normales. Algunos expertos consideran que ciertos cambios emocionales pueden atribuirse directamente al avance de la enfermedad, pero en una enfermedad tan devastadora como la ALS, puede ser difícil distinguir los sentimientos ocasionados por el trastorno subyacente, de los ocasionados por la situación de la persona.
CARACTERÍSTICAS FÍSICAS
La ELA se anuncia generalmente con una debilidad persistente o espasmos en un brazo o pierna, ocasionando dificultad para utilizar el miembro afectado, que conlleva a dificultades con estas funciones. No es poco común que las personas ignoren dichos problemas durante algún tiempo (tal vez durante meses) en esta etapa o que consulten a un médico que pueda mostrarse relativamente despreocupado.
Sin embargo, la enfermedad no se detiene ahí. Generalmente se extiende de una parte del cuerpo a otra, casi siempre a partes adyacentes entre sí, de manera que eventualmente, el problema no puede ignorarse más o tratarse mediante ejercicio o un bastón.
Entre los trastornos semejantes a la ELA se encuentran algunas formas de distrofias musculares, los trastornos neurológicos conocidos como atrofia muscular espinal bulbar y atrofia muscular espinal que aparece en adultos, el trastorno en la transmisión de nervio a músculo, conocido como miastenia grave, y diversas causas de compresión de la médula espinal o tronco cerebral, tales como tumores y malformaciones.
La pérdida progresiva de la fuerza muscular y la coordinación finalmente interfieren con la capacidad para llevar a cabo actividades rutinarias, como subir escaleras, bajarse de una silla o deglutir. Ocasionalmente, los músculos de la respiración y de la deglución son los primeros en verse afectados.
A medida que la enfermedad progresa, otros músculos resultan afectados y la persona se va incapacitando progresivamente, aunque no hay efecto sobre la capacidad para pensar o razonar.
Al comienzo de la enfermedad son frecuentes los calambres musculares, fundamentalmente por la noche, en la que los músculos tienen tendencia a contraerse espontáneamente.
La característica más sobresaliente es la debilidad progresiva en una persona que presenta atrofia muscular con hiperreflexia (exageración de los reflejos), fasciculaciones (movimientos finos o crispación muscular de una pequeña área de músculo) y calambres musculares y no se encuentra alterada la sensibilidad (ausencia de síntomas sensitivos).
Los síntomas son variables en cada persona afectada.
Suele comenzar con:
- Sensación de cansancio general.
- Pequeños temblores musculares bajo la piel (fasciculaciones); calambres.
- Torpeza en alguna extremidad.
- Hiperreflexia (exageración de los reflejos).
EVOLUCIÓN DE LA ENFERMEDAD
Las manos y los pies son las partes del cuerpo que antes suelen afectarse, por la debilidad muscular por lo que aparecen:
- Dificultades al andar.
- Dificultades en otras actividades cotidianas: lavarse, vestirse, etc.
- La parálisis puede extenderse a los músculos del cuello y tronco.
- Problemas al tragar (disfagia).
- Problemas al masticar.
- Problemas al respirar.
Puede haber espasticidad generalizada provocando risas y llantos inapropiados, que son parte de la enfermedad y no una alteración mental.
La Esclerosis Lateral Amiotrófica sólo ataca a las células motoras, por lo que los sentidos no se ven afectados:
- No hay alteración de la vista, olfato, gusto y tacto.
- No hay alteración de los esfínteres de la vejiga o el recto.
- No hay alteración de los músculos de los ojos.
- Permanecen intactas las funciones musculares automáticas como el corazón y los intestinos.
- La función sexual no se ve afectada.
- La enfermedad por sí misma no produce dolor.
- Hay que tener en cuenta que las funciones mentales no se ven alteradas.
Se distinguen tres formas clínicas:
- Común: suele iniciarse de forma asimétrica por una extremidad superior.
- Pseudopolineurítica: se inicia por las extremidades inferiores también de forma asimétrica y va progresando de forma ascendente lentamente.
- Bulbar: evolución más rápida y prácticamente desde el principio hay disfasia (dificultades en el habla), disfagia (dificultades para tragar), salivación excesiva y aumento de las mucosidades.
Los síntomas adicionales que pueden estar asociados con la enfermedad son:
- Aumento de la frecuencia/urgencia urinaria
- Espasmos musculares
- Contracciones musculares
- Atrofia muscular
- Edema en tobillos, pies y piernas
- Pérdida de peso
- Babeo
CARACTERÍSTICAS DEL LENGUAJE
- Cuando se ven afectados los nervios que controlan los músculos del habla (fonatorios) se produce una voz de tipo nasal que en ocasiones es el primer síntoma de la enfermedad
- Dificultad de los músculos que controlan el habla y la deglución
- El paciente presentara trastornos de articulación de los fonemas
- Presentara Disartria y la perdida del lenguaje hasta llegar a ser incomprensible
- También presentara Disfagia y sialorrea
La disfagia debida a los disturbios de motilidad de la lengua, faringe y esófago pueden llevar a atragantamientos y aspiración, especialmente con líquidos y comidas que se desmoronan. Así el primer paso y uno de los objetivos en el manejo nutricional, es el cambio de la consistencia de la dieta (la comida debe ser fácil de tragar y que prevenga el riesgo de aspiración, así como evitar alimentos con sabor y temperatura extremos porque aumentan la salivación), efectuar pequeñas comidas incrementando su número, masticar lenta y cuidadosamente, alimentación con el tronco erguido y la cabeza ligeramente flexionada, concentración en el acto de la deglución procurando no hablar; además proporcionar una dieta rica en energía, fluidos, vitaminas y minerales necesarios para el paciente. Se les debe explicar a los familiares como llevar a cabo la maniobra de Heimlich en caso de atragantamiento. Las técnicas especiales para la salivación como la salivación supraglótica pueden ser enseñadas por el terapista de lenguaje o el médico de rehabilitación ya que reducen el riesgo de aspiración. Generalmente después de que se establecieron estas medidas, el aporte calórico es insuficiente y el paciente inicia con una importante pérdida de peso, y se agrega que la ingesta oral de alimentos se vuelve intolerable debido a la frecuencia de atragantamientos, y es cuando la enterogastrostomía percutánea o la miotomía del músculo cricofaríngeo deben ser discutidas. La realización de la enterogastrostomía percutánea se puede hacer con anestesia local sin mayores complicaciones en fases tempranas de la enfermedad. Sus principales beneficios son su fácil inserción, la flexibilidad del tipo de dieta, su infusión por medio de bolos o continua y el riego bajo de diarrea o calambres en el caso de una yeyunostomía. Si se retrasa su aplicación hasta la etapa terminal en la cual el paciente presenta dificultad respiratoria, el procedimiento puede ser peligroso por el riesgo de atelectasia basal. Debe recordarse que aunque se realice la enterogastrostomía percutánea, el riesgo de neumonía por aspiración está latente.
La sialorrea es también frecuente; el uso de medicamentos puede reducirlo). Si la secreción es espesa, la N-acetilcisteína puede ser usada, junto con una ingesta de líquidos suficiente. Los inhaladores y la succión son necesarios; los betabloqueadores pueden ser una alter- nativa en los casos severos. La irradiación de la glándula salival y la neurotomía transimpática han sido exitosas en reportes anecdóticos
TRATAMIENTO TERAPIA DE LENGUAJE
Los primeros estudios realizados en relación con la comunicación y el lenguaje en pacientes con Esclerosis Lateral Amiotrófica, centraban su interés en el comportamiento motor del lenguaje, es decir, en el habla. Esta tendencia tiene su explicación en el hecho de que se consideraba que estos pacientes sólo presentaban síntomas predominantemente motores. Por ello, muchas investigaciones se centraron en evaluar los componentes articulatorios, fonatorios y respiratorios del habla, mencionando dos tipos básicos de trastornos: las disartrias que son trastornos de articulación, y las disfonías, que son trastornos de la fonación.
Puesto que se trata de personas con un deterioro neuromotor degenerativo generalizado, los problemas de comunicación tienen una base biológica que no sólo se plasma en las limitaciones de producción del lenguaje, es decir del habla; sino que comprometen también el aspecto motor en general, y repercute seriamente en aspectos paralingüísticos como pueden ser las expresiones faciales, el movimiento corporal, los gestos, etc.;
Otro elemento a destacar es que algunas evaluaciones neuropsicológicas en pacientes de ELA han puesto en evidencia que algunos de los afectados presenten deterioros cognitivo y del lenguaje, que comprometen su comunicación, de manera drástica. A nivel cognitivo se han descrito cambios profundos de personalidad, desinhibición, jocosidad, etc.; en el lenguaje se han descrito reducciones paulatinas de su producción, errores persistentes, dificultades en resolver tareas de nominación y en la comprensión de frases complejas etc. Todo lo anterior, indica que el proceso patógeno de la ELA es más extenso de lo que se pensaba anteriormente. Siendo todos estos aspectos graves y comprometedores de la capacidad de comunicación, el más importante sin duda es la falta de deseo o de interés por comunicar.
Normalmente sucede en las fases finales de la enfermedad, a no ser que se trate de una ELA bulbar, en el que este síntoma se da desde el principio (junto con las dificultades para tragar, excesiva salivación y moco).
Es conveniente conservar la energía, hablar lentamente, con frases cortas y utilizar las señales no verbales, intentando utilizar frases fáciles de pronunciar.
Perder la capacidad de hablar correctamente supone un cambio muy significativo en la persona; puede ser frustrante tanto para el paciente como para el interlocutor que, inconscientemente, se lo transmite a él. La persona tiende a suprimir las intervenciones espontáneas y participar en conversaciones. Es útil saber qué músculos se encuentran afectados y cómo hacer el mejor uso de ellos.
Existen en la actualidad multitud de aparatos en el mercado que le ayudarán a mantener el contacto con las otras personas y compartir la información. El equipo de comunicación que usted necesite es, en muchas ocasiones, una decisión personal; mientras algunas personas evitan un equipo electrónico de comunicación, otras requieren el más sofisticado. Se debe elegir el equipo según las necesidades individuales en el momento y en el futuro.
Hay algunos factores que pueden ser relevantes a la hora de elegir un equipo de comunicación:
- Nivel de entrenamiento necesario y la complejidad del sistema.
- Coste.
- Facilidad de transporte.
- Flexibilidad.
- Velocidad de comunicación.
- Capacidad de memoria.
Tabla 1 APOYOS TÉCNICOS A LA COMUNICACIÓN
| GRADO DE AFECTACIÓN |
CARACTERÍSTICAS GENERALES |
APOYOS TÉCNICOS |
|
LEVE |
Disartria leve a moderada. Sin compromiso o ompromiso leve en miembros superiores e inferiores. |
Uso de cuaderno o pizarra |
|
MODERADO |
Disartria moderada a severa. Sin compromiso o compromiso leve a moderado de miembros superiores e inferiores. |
Uso de pizarras programas para ordenador: PREDICE; tableros de comunicación: alfabético y pictogramas; Comunicadores: CANNON y LIGHTWRITTER |
|
SEVERO |
Anartria. Compromiso severo en miembros superiores e inferiores. |
Programas para ordenador y tableros de comunicación (alfabético, pictogramas). |
La presente tabla indica el grado de afectación de la ELA y las estrategias y apoyos técnicos que se pueden utilizar, de acuerdo con los recursos existentes, en base a las evaluaciones realizadas por profesionales de la Comunicación. Como puede observarse, uno de los últimos recursos de apoyo a la comunicación se basa en la utilización de programas de ordenador. De ahí la importancia de que estos pacientes se familiaricen con su uso.
El que puedan usar ordenadores los pacientes con un grado de afectación severo, no significa que no deban y puedan hacerlo en la fase leve sino todo lo contrario.
La disartria y la pérdida del lenguaje hasta llegar a ser incomprensible, deben ser manejadas para darle al paciente opciones de comunicación. Ante todo es importante una gran paciencia y concentración para comprender al paciente. Para ayudar al habla pueden estimularse los músculos faciales pasando un cubito de hielo alrededor de los labios. Así mismo, la valoración del terapista de lenguaje puede permitir hacer lo más posible por la capacidad restante del habla y los familiares pueden pasar mucho tiempo estimulando la comunicación. Pueden usarse como medidas auxiliares: desde una simple tabla o pizarrón con gis o pluma, tablero con palabras para que el paciente las señale, una máquina de escribir, o hasta aparatos sofisticados computacionales en casos de plejía total manejados por medio de computadoras personales con acceso a Internet, controles ambientales, controles biomioeléctricos sensibles y/o activados por medio de la voz.
En ELA podemos agrupar el trabajo del Logopeda en torno a tres campos o momentos:
- Preventivo
Una vez hecho el diagnóstico podemos prever la evolución futura de la enfermedad .Por ello es aconsejable iniciar la terapia incluso antes de la instauración de los síntomas, con vista de evitar un deterioro superior a lo deseable. En este momento lo que pretendemos es conservar la movilidad cervical y facial (especialmente la de los órganos que intervienen en el habla y la deglución)
Mantener la capacidad respiratoria ya que la perdida de la función del aparato respiratorio puede pasar desapercibida en reposo y solo ponerse de manifiesta cuando se realiza ejercicio físico .Así que si limitamos el ejercicio puede pasar desapercibido durante mucho tiempo hasta que se haya perdido toda la capacidad de reserva.
- Rehabilitador
Cuando ya empiezan a aparecer los síntomas actuaremos sobre los mismos para evitar una progresión de los déficit superiores a los deseados .Así que haremos ejercicios de movilidad cervical y de praxis bucofonatoria.
Ejercicios de respiración, con el fin de aumentar los tiempos respiratorios.
Ejercicios de fonación a través de la emisión de vocales, después silabas, palabras y frases siempre controlando la emisión del aire.
Ejercicios de articulación mediante actividades de lectura en voz alta con articulación forzada y controlada.
- Compensatorio.
Cuando se han agotado todos los tratamientos rehabilitadotes y las posibilidades de omisión de voz funcional son escasas y nulas, es preciso buscar métodos alternativos que faciliten la comunicación. Podemos encontrar distintos niveles dependiendo de la afectación que en orden de progresión son los siguientes.
La escritura, con adaptaciones, quedando la opción de utilizar comunicadores u ordenador.
Tableros de comunicación señalando a través de un puntero u otros medios cuando hay severos problemas de los miembros superiores.
Ordenador con adaptaciones para el pulsador
TRATAMIENTO DE TERAPIA FÍSICA:
La debilidad es la principal queja de los pacientes con ELA. La fisioterapia activa y pasiva es importante, debido a que previene las contracturas musculares y la rigidez articular. La cantidad y el tiempo de ejercicio hecho por el paciente pueden variar de un día a otro; como regla el paciente no debe nunca hacer ejercicio al punto de quedar exhausto; se recomienda incorporar periodos de descanso durante la terapia y la rutina diaria. Si las piernas están severamente afectadas y hay un elevado riesgo de caídas, la terapia en la alberca puede ser de ayuda. A medida que la enfermedad avanza, el paciente requiere de aparatos adicionales para mantener la movilidad (como férulas en tobillos o silla de ruedas) y para mantener la independencia en las actividades de la vida diaria (instrumentos especiales para comer, taza de baño elevada, tubos empotrados en la pared al lado de la tina de baño, etc.). Es importante hablar con el paciente sobre la necesidad de estos aparatos, ya que en muchas ocasiones el uso de los mismos es rápido y progresivo y puede que el paciente y su familia, no tengan el tiempo necesario para ajustarse psicológicamente a dichos cambios. La evaluación del terapista ocupacional es necesaria en este punto para brindar los medios necesarios: aparatos para el vestido, aditamentos en el baño, etc. Los inhibidores de la acetilcolinesterasa pueden ser usados por cortos periodos para mejorar la fuerza muscular, especialmente durante las fases iniciales de la enfermedad. Su efecto es más pronunciado en pacientes con síntomas bulbares. Sin embargo, este tratamiento no es efectivo en todos los pacientes, y sólo dura algunos días o semanas. Se recomienda el uso de piridostigmina (40 mg) para situaciones especiales, como un viaje o una fiesta.
La presencia de fasciculaciones y de espasticidad, puede ser tratada por medio de medicación apropiada. En el caso de las fasciculaciones el uso de sulfato de quinina, carbama- zepina, vitamina E, fenitoína, magnesio o verapamilo, se ha reportado con utilidad para estos pacientes. Para la espasticidad, el uso de baclofen, tizanidina o tetracepan han mostrado buenos resultados.
Rehabilitación
En el tratamiento de la persona afectada por la enfermedad es conveniente la participación de un equipo multidisciplinar en el que se encuentren el neurólogo, rehabilitador, fisioterapeuta, logopeda, terapeuta ocupacional, dietista, neumólogo, digestivo, personal de enfermería, psicólogo y asistente social. Cada persona afectada es única y las mismas lesiones evolucionan de manera diferente de una persona a otra, por lo que es necesario un tratamiento individualizado, revisado y adaptado periódicamente.
Los objetivos generales que debería perseguir un programa de rehabilitación son los siguientes:
- Educación sanitaria.
- Prevenir deformidades.
- Uso sensato de la energía.
- Normalizar el tono muscular.
- Mantener los mecanismos posturales normales.
- Mejorar la coordinación, equilibrio y estimular la marcha.
- Mantener la amplitud articular.
- Estimular toda experiencia sensitiva.
- Ergonomía postural en cada fase de la enfermedad (la ergonomía es el estudio de datos biológicos y tecnológicos aplicados a problemas de mutua adaptación entre el hombre y la máquina).
- Integrar los ejercicios en AVD (Asistencia Ventilatoria Domiciliaria).
- Apoyo psicológico.
Los procedimientos de rehabilitación serán diferentes dependiendo de la etapa de la enfermedad y las limitaciones de la persona.
Etapa de Independencia
El paciente es ambulatorio y se manejó sólo en la AVD. Sólo hay debilidad leve y/o torpeza muscular.
Si hay debilidad
- Técnicas del concepto Bobath. El concepto Bobath se basa en dos principios fundamentales:
- Inhibir el tono anormal mediante la utilización de posturas que lo disminuye.
- Facilitar las reacciones automáticas deseadas.
- Fundamentos del Concepto Bobath:
- Enfatizar sobre la calidad del movimiento.
- Buscar resultados de larga duración en pacientes ortopédicos especialmente.
- El programa de abordaje se planifica para cada individuo.
- El paciente debe ser visto como un todo.
- El entrenamiento a los padres es esencial para el manejo de los niños.
- Técnicas de FNP método Kabat: (facilitación neuromuscular propioceptiva):
- Se basa en el principio de que todo acto motor es una elaboración del SNC, en respuesta a una múltiple información sensitivo - motora simultánea y secuencial, de manera que puede influirse o modificarse mediante diversos estímulos táctiles, auditivos, visuales, etc.
- Primeramente se fatiga al músculo para relajarlo. Al estirar se contraen los músculos contrarios (antagonistas) causando algo conocido como inhibición recíproca, esto hace que se relaje el músculo que se desea relajar.
- Masoterapia estimulante: mediante la masoterapia (masajes) se eliminan las sustancias sobrantes del músculo y facilita su irrigación y reposición después de una contractura muscular.
- Masaje relajante general o en regiones.
- Masaje centrípeta.
- Masomanipulación por regiones.
- Drenaje postural y terapia percutiva.
- Cinesiterapia activa libre y resistida
En caso de espasticidad:
- Técnicas de inhibición de Bobath.
- Técnicas de relajación.
- Movilizaciones pasivo – suaves y activo – asistidas.
- Estiramientos analíticos y globales.
- Miembro inferior: músculos posteriores del muslo, flexores y aproximadotes de cadera
- Miembro superior: rotadores del hombro, flexores del codo, pronadores anteriores del antebrazo (músculos que vuelven la palma de la mano hacia abajo), flexores de muñeca y dedos y flexores laterales del tronco.
- Masoterapia descontracturante: masaje manual.
- Hidroterapia y natación con ejercicios dirigidos.
Para mejorar el equilibrio y la coordinación
- FNP/Kabat: estabilizaciones rítmicas, ejercicios en colchoneta, volteos, etc.
- Ejercicios en cuadrupedia (las cuatro extremidades apoyadas en el suelo).
- Ejercicios de coordinación de Frenkel. Consisten en una serie de ejercicios que pretenden hacer emplear al paciente lo que se conserva de su sentido muscular con el objetivo de evitar su disminución progresiva e incluso conseguir una mejoría.
- Los ejercicios deben empezar con movimientos muy simples y deben progresar gradualmente hasta los más complicados.
- Cada ejercicio o grupo de ejercicios debe ser capaz de realizarlo en forma correcta antes de permitirle pasar a un ejercicio más difícil. Debe lograr una suficiente precisión, pero los ejercicios deben ser suficientemente variados para evitar el aburrimiento.
- No deben realizarse ejercicios que supongan un intenso trabajo muscular. La progresión se realiza por complejidad, pero no por potencia.
- Ningún movimiento debe sobrepasar su límite normal, ya que la hipotonía de los músculos y la laxitud de los ligamentos puede predisponer a la luxación.
- Los movimientos deben realizarse, al principio, más bien en forma rápida y después más lentamente; esta última modalidad es más difícil ya que exige una mayor precisión.
Reeducar y conservar la marcha
- Mediante FNP (Facilitación Neuromuscular Propioceptiva)
- Ejercicios funcionales.
- Distintos tipos de marcha: hacia delante, hacia atrás, lateral, etc.
- Marcha asistida.
- Hidroterapia en piscina.
Estimular la práctica deportiva
- Mediante ejercicios aeróbicos y de alto nivel de resistencia, como la natación, marcha rápida, etc.
- Enseñanza y supervisión de un programa de ejercicios diarios en domicilio.
- Etapa de debilidad moderada
- A los ejercicios anteriores se le suma:
- Movilizaciones asistidas y resistidas
- Uso de mecanoterapia adaptada
- Empleo de ortesis (aparatos para suplir la debilidad de los músculos que flexionan el pie o extienden la muñeca)
- Inicio de un programa de rehabilitación respiratoria
Etapa de debilidad severa
A lo anterior se le suma:
- Movilizaciones activas asistidas o pasivas y ejercicios de musculación isométricos.
- Prevención del hombro doloroso.
- Ejercicios para la musculatura facial.
- Hidroterapia.
- Intensificar la fisioterapia respiratoria.
- Adaptaciones del hogar.
Etapa de "silla de ruedas"
- Fisioterapia respiratoria.
- Cambios frecuentes de postura y corrección postural.
- Mesoterapia circulatoria.
- Independencia en silla de ruedas.
- Ergoterapia: economía del trabajo.
- Verticalización diaria.
Etapa "en cama"
A todo lo anterior se le suma:
- Cuidados posturales.
- Fisioterapia respiratoria:
- Ventilación dirigida.
- Tos asistida.
- Percusión.
- Vibración suave.
- Drenajes posturales.
- Uso del aspirador.
- Adiestramiento en el uso de mascarillas a la familia o cuidador.
- Movilizaciones pasivas o activo – asistidas según estado de la persona.
- Drenaje linfático manual.
- Medias elásticas.
Ventilación Mecánica a Domicilio (VMD)
Hay que tener en cuenta que la causa más frecuente de muerte es por insuficiencia respiratoria por disfunción de los músculos respiratorios. Los signos y síntomas de fallo respiratorio suelen presentarse meses o años después de los síntomas iniciales de debilidad. Por la rápida evolución de la enfermedad, es difícil tomar la decisión de someterse a ventilación mecánica domiciliaria.
Este soporte ventilatorio se realiza con unos aparatos (cada vez más perfeccionados, más manejables, etc.) que suministran un volumen de aire o volumen corriente, a una frecuencia que concede al paciente un tiempo para inspirar y otro para expirar. Desde hace tiempo, esta VMD se realiza de forma no invasiva por medio de una mascarilla nasal, pero existen algunas limitaciones para su uso. Se debe intentar la ventilación no invasiva cuando:
- El paciente está consciente.
- Es capaz de toser eficazmente.
- Progresión lenta de la enfermedad.
- Conserva cierto grado de autonomía respiratoria.
- Pacientes muy motivados y con grandes deseos de vivir.
- La persona tiene capacidad de comunicarse y desarrollar alguna de las actividades de la vida normal.
- consigue, para evitar que en un futuro afloren inútiles sentimientos de frustración y pena que pueden tener una influencia negativa sobre el pronóstico.
- El equipo profesional tiene experiencia.
Si no se cumplen estas condiciones hay que practicar una traqueotomía (o traqueostomía), operación que consiste en una incisión en la tráquea para formar una abertura por el que se introduce un tubo).
Los pacientes también pueden presentar problemas derivados de un mal drenaje de las secreciones, que es muy frecuente cuando hay afectación bulbar progresiva, por lo que es muy eficaz los métodos de tos asistida y drenaje de las secreciones.
Algunos autores proponen comenzar la Ventilación Mecánica de manera precoz para que la persona se sienta familiarizada con el equipo cuando sólo sería necesaria una utilización parcial y así evitar las muertes prematuras en pacientes de evolución rápida y estar preparados para cuando sea necesario un soporte completo. De todas maneras, otros autores sostienen que es mejor comenzar cuando el paciente presente las primeras manifestaciones de insuficiencia respiratoria aguda.
Un aspecto en el que hay consenso total es en la capacidad de prolongar la vida de la Ventilación Mecánica no Invasiva.
Se debería optar por la traqueotomía cuando:
- Hay problemas importantes de limpieza de secreciones.
- Tos ineficaz.
TERAPIA OCUPACIONAL
La Terapia Ocupacional supone el tratamiento y adiestramiento del paciente de modo que alcance su máxima capacidad para realizar una vida lo más normal e independiente posible desde el punto de vista físico, psíquico, social y laboral, optimizando la capacidad del paciente, minimizando sus déficit y teniendo en cuenta su forma de vida y el entorno socio-familiar que le rodea.
INTERVENCIÓN DESDE LA TERAPIA OCUPACIONAL
El terapeuta ocupacional cuando recibe a un paciente con diagnóstico médico , hará una evaluación que puede tener varios objetivos dependiendo de la razón por la cual se envía al paciente a terapia ocupacional.
Las razones más comunes para llevar a cabo una evaluación son:
- Efectuar un examen de la discapacidad, el propósito es identificar a los pacientes que tienen discapacidades (disfunciones en el desempeño de las tareas) y que son susceptibles de una evaluación más extensa. Se efectúa una evaluación al ingreso de la asistencia psiquiátrica.
- Es deseable tener procedimientos de selecciones breves y fáciles de administrar. Deberán ser muy sensibles.
- Concretamente preguntar al paciente acerca de:
- ¿Cómo describiría sus perspectivas en la vida?
- ¿Tiene dificultad para dormirse? ¿Cuántas veces se despierta a lo largo de una noche? ¿Cuánto tiempo tarda en volver a dormirse?
- ¿Qué actividades le producen placer últimamente? ¿Qué actividades ha abandonado últimamente?
- ¿Cómo se siente usted últimamente? ¿Cree que los demás le valoran? ¿Siente usted que es una carga o que es una mala persona?
- ¿Ha disminuido su energía en los últimos tiempos? Hábleme de sus actividades diarias. ¿Qué hace por la tarde?
- ¿Ha notado algún cambio en su capacidad de concentrarse? ¿Ha notado algún cambio en su memoria?
- ¿Qué tal está de apetito? ¿Ha variado su peso?
- ¿Se siente triste a menudo? ¿Ha pensado en hacerse daño a usted mismo?
- Describir el estado funcional.
Se realiza para proporcionar información acerca de las tareas de la vida diaria que puede desempeñar el paciente de forma independiente y dependiente.
- Proporcionar datos para planificar una intervención de terapia ocupacional.
Para planificar un programa individualizado de terapia ocupacional es importante hacer una evaluación. Una evaluación de tratamiento para planificar la intervención va más allá de lo necesario para describir un estado funcional. Además de determinar el nivel de independencia para el desempeño de las tareas, el terapeuta también debe determinar el potencial del paciente para mejorar o recuperar el estado funcional y el medio por el cual se puede lograr este cambio. Esto exige que el terapeuta evalúe la capacidad de un paciente para aprender o mínimamente responder a las modificaciones ambientales.
En otras palabras, no es suficiente con que un terapeuta determine si un paciente es dependiente en la tarea de bañarse. El terapeuta también debe determinar si el paciente es capaz de cambiar de un estado dependiente a otro más independiente en el baño.
Un banco para traslados en la bañera permitió a la Sra. X superar su temor a caerse en la bañera; por ende ella pudo reanudar los baños. No obstante cuando se utilizó la misma intervención en la Sra. Y, su preocupación de deslizarse se trasladó del piso de la bañera a la silla y siguió siendo dependiente para el baño.
- Ayudar a determinar la competencia para la vida independiente.
Los déficit en las tareas de movilidad, cuidados personales y manejo del hogar,. Uno de los usos de la valoración del estado funcional es ayudar a tomar decisiones acerca de la residencia comunitaria o la colocación en lugares supervisados.
- Evaluar la necesidad de contenciones físicas.
Las sujeciones se aplican a los pacientes para controlar el comportamiento interruptor como agitación, agresión, inquietud, combatividad y divagación; para proteger al paciente y al equipo del daño, para prevenir la interferencia con el tratamiento (por ejemplo, quitarse sondas nasogástricas, vesicales, etc.).
Las contenciones pueden exacerbar la confusión, aumentar la agitación, y no prevenir los accidentes. Las contenciones predisponen a los pacientes a padecer lesiones cutáneas, estreñimiento y desorientación. Las medicaciones tranquilizantes se han utilizado como sustitutas de las contenciones físicas.
Las contenciones no deben utilizarse para disciplina ni conveniencia.
Las contenciones físicas son: chalecos, guantes, cinturones de asiento, lazos para las muñecas/codos, tablas de contención y sillas geriátricas.
Las sujeciones físicas pueden ser utilizadas sólo para "conseguir la seguridad física de un paciente y sólo con la orden escrita de un médico que especifique la duración y las circunstancias bajo las cuales se van a utilizar las contenciones".
EVALUACIÓN DE TERAPIA OCUPACIONAL
La evaluación de terapia ocupacional se centra en el desempeño de las tareas de la vida diaria. Las tareas de la vida diaria abarcan cinco categorías principales: movilidad, cuidados personales, manejo del hogar, recreo y trabajo.
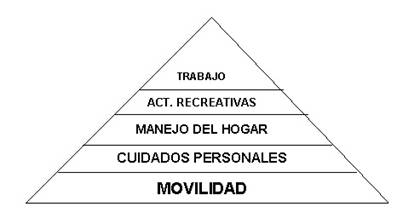
Se evalúan dos dimensiones: Destreza, que comprende la evaluación de la habilidad, o sea, lo que los pacientes son capaces de hacer en grados de independencia, seguridad y corrección que un paciente utiliza para completar una tarea. La evaluación de las destrezas informa acerca de lo que un paciente es capaz de hacer (habilidad) y de la discapacidad.
Los Hábitos constituye la segunda dimensión del desempeño de tareas que se evalúa. Se refieren a las tareas rutinarias del paciente, describe la frecuencia con que el paciente utiliza sus habilidades para hacer las tareas.
Las cinco categorías de la vida cotidiana forman una jerarquía:
MOVILIDAD
- La movilidad proporciona una impresión global del estado funcional del paciente.
- En la cama (de un lado a otro/ Hacia la cabecera y a los pies de la cama/ Hacia la posición de sentado)
- Traslados: Hacia la cama, a una silla, al retrete, a la bañera, al automóvil.
- Bipedestación
- Deambulación
- Subir y bajar escalones
- Girar
- Alcanzar algo por encima de la cabeza
- Inclinarse, agacharse
- Capacidad de moverse con propósito de un lugar a otro dentro y fuera de la casa , encontrar los caminos
- Observar si hay fatiga en la realización de estas actividades.
- (Por ejemplo, podemos observar a un paciente que muestra inestabilidad en la bipedestación y deambulación y sin embargo cuando realiza tareas de vestido o de hacer las camas está más seguro y estable).
CUIDADOS PERSONALES
Este tipo de tarea comprende la habilidad para cubrir las necesidades personales básicas y exigen que el paciente controle los objetos necesarios para realizar la tarea a la vez que controla su cuerpo.
La persona debe mantener autocuidados en:
- Alimentación
- Control de esfínteres
- Baño
- Higiene
- Arreglo personal
- Vestido
- Comunicación funcional
Por ejemplo, en la actividad de "peinarse", hay que controlar el peine, coger el peine y mantenerlo en la mano, llevarlo hasta la cabeza, realizar el movimiento del peinado.
Concretamente en la depresión tendremos en cuenta los siguientes aspectos:
- Apetito
- Pérdida de peso (o aumento)
- Incontinencia vesical e intestinal
- Olor del cuerpo
- Aspecto personal
- Esto factores citados se evalúan para tener información de los hábitos del paciente.
MANEJO DEL HOGAR
Imponen mayores demandas de desempeño funcional que las anteriores porque exigen mayor control de objetos, algunos muy sofisticados.
Factores que se evalúan:
Movilidad avanzada
- Entrar y salir del hogar con manipulación de puertas y cerraduras.
- Caminar llevando un peso (sacar la basura)
- Subir a una banqueta o escalerilla (cambiar una bombilla)
- Moverse en la comunidad (utilizar el coche o autobús)
Cuidados personales avanzados
- Manejar las medicaciones (apertura de recipientes y manejar la posología)
- Utilizar el teléfono para obtener información, hacer citas, solicitar ayuda en una emergencia)
Tareas domésticas livianas
- Ir a comprar alimentos (se puede hacer por teléfono) y ropa.
- Planificar alimentos calientes y fríos
- Lavar y secar platos
- Cuidar vestimentas ( lavado, secado, planchado, cosido)
- Aspirar, barrer el suelo
- Hacer una cama
- Realizar reparaciones pequeñas (una lámpara)
- Regar el jardín, rastrillar hojas.
Tareas domésticas pesadas
- Cambiar ropas a la cama
- Limpiar elementos de la casa (frigorífico, ventanas, baños...)
- Tareas de mantenimiento del hogar (colgar cortinas, un cuadro...)
- Quitar nieve
- Cortar césped
- Arrancar las malas hierbas
Aspecto económico
- Manejar dinero para las compras
- Pagar facturas
- Rellenar cheques y controlarlos
Está claro que el manejo del dinero deficiente es incompatible con la vida independiente.
El observar el frigorífico con el alimento derramado, las habitaciones desordenadas, la acumulación de basura, etc. informan acerca de los hábitos del paciente.
ACTIVIDADES RECREATIVAS
El terapeuta ocupacional en este aspecto debe conocer:
- Los gustos e intereses del paciente
- Las destrezas
- La participación
La participación recreativa en la vejez da estructura y sentido a los años posteriores a la jubilación y es el vehículo para mantener la agudeza física mental y social.
Pueden ser graduadas dependiendo de las demandas:
- Motoras
- Cognitivas
- Emocionales
Ejemplos de actividades son: Trabajo artístico, jugar en solitario (requiere un menor esfuerzo que jugar en grupo), Jugar en grupo, juego en equipo (bolos).
HABILIDADES DE ORGANIZACIÓN
Es necesario que el paciente tenga capacidad de organizarse el día para integrar las tareas cotidianas en los hábitos y mantener un estilo de vida eficaz.
Los pacientes con depresión pueden mostrar destrezas para realizar las tareas del hogar pero no pueden planificar ni cumplir un esquema de vida organizado.
MÉTODOS PARA EVALUAR LOS HÁBITOS DEL PACIENTE
OBSERVACIÓN
Es el método más eficaz, pero muchas veces imposible realizarlo.
Cuando observemos al paciente realizando la tarea debemos prestar atención a:
- Grado de ansiedad
- Agitación psicomotora
- Retraso psicomotor
- Temblor
- Somnolencia
- Impulsividad
- Síntomas somáticos
- Alucinaciones o delirios.
ENTREVISTA
Es el método más utilizado, se necesita la presencia de un familiar del paciente para obtener datos reales, el familiar intervendrá exclusivamente cuando se lo pidamos.
De la observación el terapeuta ocupacional obtendrá en diagnóstico funcional, por ejemplo, si un paciente presenta una disfunción en el vestido habrá que valorar qué es lo que impide la realización de la tarea, puede haber una desmotivación por la depresión que presenta (trastorno afectivo) pero además puede presentar una limitación de la flexión de cadera. Deberemos tener en cuenta ambas deficiencias a la hora de planificar el tratamiento.
PARTICIPACIÓN EN ACTIVIDADES SIGNIFICATIVAS.
Se trata de hacer participar a los pacientes en un número limitado de tareas en un tiempo determinado. Las tareas deben ser compatibles con las destrezas del paciente así como de sus intereses.
Por ejemplo, se podría enseñar a un paciente como doblar y colocar su ropa en el armario y de esta forma facilitarle la tareas del vestido cuando vaya al armario. También le podríamos indicar que llevara su ropa sucia cuando vaya de camino al desayuno.
Después de realizada la tarea, el paciente y el terapeuta ocupacional deben evaluar los puntos objetivos - cantidad y calidad del trabajo - y los puntos subjetivos - percepción y sentimiento.
El terapeuta ocupacional proporcionará retroalimentación de la realidad de los pacientes prestando un tono positivo a la evaluación.
Poco a poco se irán agregando tareas que están determinadas por la situación vital del paciente.
Por ejemplo, si el paciente va a tener que hacerse el desayuno cuando sea dada de alta, deberá ser incluida esa tarea en el entrenamiento.
Se debe tener en cuenta la fatiga que puede presentar el paciente en la realización de la tarea, por lo tanto habrá que establecer tiempos de descanso intercalados con la actividad o alternar actividades que requieran mayor esfuerzo con otras que sean menos exigentes.
El objetivo es estimular en el paciente el sentido de autocontrol.
La etapa final del entrenamiento de los hábitos consta de la transferencia del desempeño de las tareas del lugar controlado de terapia ocupacional a la circunstancia vital real. En esta etapa los pacientes deben adquirir mayor responsabilidad en los aspectos de planificación y organización.
El manejo del tiempo incluye tener en cuenta todas las tareas obligatorias, permitir las tareas recreativas y manejar acontecimientos imprevistos.
INTERVENCIONES BASADAS EN EL AMBIENTE
La capacidad para realizar las tareas tiene dos aspectos o factores:
1. Factores internos que se denominan competencias.
2. Factores externos que suponen las demandas de las tareas.
Cuando las competencias del paciente no pueden ser mejoradas, a menudo se pueden reducir las demandas para permitir una mayor participación del anciano.
Por lo tanto los objetivos perseguidos son:
- Apoyar la competencia funcional de la persona.
- Proporcionar la estimulación apropiada.
- Promover la seguridad.
La intervención basada en el ambiente incluye:
- Manipulación de estructuras construidas como cambiar escalones por rampas.
- Manipulación de objetos como incluir ayudas técnicas, quitar alfombras, modificar el sitio de los muebles, etc.
En resumen se trata de aportar al paciente un entorno seguro y cómodo donde pueda llevar a cabo una vida agradable.
RECOMENDACIONES
Paciente ELA que comienza a presentar alteraciones del habla, debe acudir a un especialista (logopeda o foniatra). No obstante, le pueden resultar de utilidad las siguientes recomendaciones:
- Utilizar frases cortas eliminando palabras innecesarias.
- Hablar despacio
- Aprender un ritmo de respiración y de pausas adecuado.
- Dar pistas al oyente mediante la expresión facial y corporal.
- Usar lápiz y papel siempre que le sea posible.
- Mantener una actitud tranquila y relajada a la hora de hablar, procurando un ambiente silencioso.
- No beber ni comer ni beber mientras habla para evitar el riesgo de aspiración.
Cuando la comunicación se hace difícil será necesario utilizar métodos alternativos, teniendo en cuenta que es necesario seleccionar el método más adecuado para cada paciente.
Muchos condicionantes y limitaciones pueden superarse si se mantiene el deseo de comunicar.
El aumento de secreciones bronquiales y de saliva puede suponer un gran problema. Si la persona se encuentra acostada, al toser no consigue expulsar estas secreciones, que se acumulan. Existen métodos para expulsar las secreciones con el cuerpo en posición lateral con los que se debe familiarizar y que le podrá entrenar con el fisioterapeuta.
Se recomienda que el paciente utilice la ventilación no invasiva para compensar por los músculos debilitados, permitiendo que el aire entre y salga de sus pulmones como si sus músculos trabajaran bien .En la ventilación no invasiva, no se hacen incisiones o invasiones corporales.
La ventilación No Invasiva se presenta en muchas formas, pero consiste generalmente en dos elementos básicos: un dispositivo interfacial, tal como una mascarilla o insertos nasales y aire suministrado a presión por una máquina portátil pequeña.
Otra forma de apoyo respiratorio, conocido como ventilación invasiva, suministra aire mediante un agujero en la tráquea.
Se debe trabajar de forma interdisciplinaria con el paciente, el fisioterapeuta el logopeda y el terapista ocupacional, y sobre todo la familia tratando de mejorar el aspecto físico, social y emocional del paciente
Los terapeutas del habla pueden sugerir que una persona con ELA grabe lo que hable .Mas tarde pueden programarse algunas frases en una computadora a la vez que la persona desearía hablar respecto de su vida, para que sus amigos y familiares lo escuchen en el futuro.
Escuchar y apoyar tanto al paciente como a su familia es una parte importante de la terapia. Todos deben ser comprometidos en la atención y en cualquier plan que se formule
El estreñimiento es un problema que se da con cierta frecuencia debido a la pobre ingestión de agua o fibra. Es tan importante una dieta rica en frutas, verduras, legumbres, hortalizas y cereales, como tomar suficientes líquidos: de litro a litro y medio de agua al día, entre comidas a ser posible, para no alargar la digestión.
La mejor manera de combatir la fatiga es conservar la energía para las tareas o actividades que realmente sean importantes y gratificantes para usted. El terapeuta ocupacional puede planear unas actividades diarias que le ayuden a adaptarse a la vida diaria con la enfermedad. Existen gran cantidad de ayudas para hacerle la vida menos difícil, pero consulte primero con su terapeuta su problema en particular
CONCLUSIONES
ELA es una de las enfermedades neuromusculares más comunes en el mundo entero y afecta a persona de todas razas, generalmente se da entre los 40 y 60 años de edad, pero también la pueden desarrollar personas jóvenes .Los hombres son mas afectados a menudo que las mujeres
En el 90% a 95% de los casos de ELA la enfermedad ocurre aparentemente en forma aleatoria sin ningún factor de riesgo claramente asociado .Los pacientes no tienen una historia familiar de la enfermedad y no se considera que los miembros de su familia tengan un riesgo mayor de desarrollar ELA.
Entre el 5% y el 10% de todos los casos de ELA son heredados .La forma familiar de ELA generalmente resulta de un patrón hereditario que requiere que solamente uno de los padres lleve el gen responsable por la enfermedad, es claro que existen otras causas genéticas, no identificadas.
Las dificultades del habla se presentan en un 75% de los pacientes.
Las anormalidades tales como la disfonía y la disartria son los síntomas tempranos más comunes de la forma bulbar del comienzo.
El logopeda debe buscar, con ayuda del paciente y sus familiares, los métodos más eficaces y sencillos que aseguren la comunicación entre el paciente y su medico para evitar aislamiento y asegurar su perfecta atención.
Se puede utilizar estrategias simples para mejorar la alimentación. Un collar cervical ayuda a corregir la debilidad de los extensores del cuello y cambios de la dieta pueden facilitar la deglución
BIBLIOGRAFÍA
- REVISTA MEXICANA DE REHABILITACION FISICA Y REHABILITACION 2003
- MANUAL DE CUIDADOS PARA PERSONAS AFECTADAS DE ESCLEROSIS LATERAL AMIOTRÓFICA- ADELA Asociación Española de Esclerosis Lateral Amiotrófica
- Enciclopedia médica Microsoft - Encarta 2004