
Mª TERESA JIMÉNEZ GARCÍA
FISIOTERAPEUTA POR LA UNIVERSIDAD DE SALAMANCA
FISIOTERAPEUTA COLEGIADO CL05-0340
ÍNDICE
RESUMEN
PALABRAS CLAVE
1. INTRODUCCIÓN
2. MARCO TEÓRICO
2.1. Definición
2.2. Clasificación
2.3. Epidemiología
2.4. Etiología
2.5. Diagnóstico
2.6. Manifestaciones clínicas
2.7. Tratamiento
2.7.1. Farmacológico
2.7.2. Fisioterápico
2.8. Escalas de Valoración
3. MATERIAL Y MÉTODO
3.1. CASO CLÍNICO
3.1.2. Antecedentes personales
3.1.3. Antecedentes familiares
3.1.4. Exploración inicial
3.1.5. Objetivos y tratamiento
3.1.6. Resultados
4. CONCLUSIONES
BIBLIOGRAFÍA
RESUMEN:La amiotrofia espinal infantil es una enfermedad autosómica recesiva que se caracteriza por la degeneración de las motoneuronas alfa de la médula espinal. Se ha clasificado en tres tipos, existiendo además una forma tardía que aparece en la edad adulta. Tiene una incidencia de entre 1/6.000 y 1/10.000 de los nacidos vivos. Se produce por una alteración en el brazo largo del cromosoma 5, locus D5s39. Las manifestaciones clínicas se caracterizan por debilidad muscular generalizada, arreflexia osteotendinosa, hipotonía y atrofia muscular proximal simétrica. En la actualidad, no existe un tratamiento que remedie la enfermedad, aunque se están realizando estudios en medicamentos para retrasar la progresión de la misma. El tratamiento de fisioterapia principalmente es de sostén, paliando las complicaciones que conlleva la progresión de la enfermedad.
En el presente artículo se describe un caso clínico de amiotrofia espinal infantil. El paciente fue diagnosticado cuando tenía dos años de edad, tras lo cual se realizó un estudio genético de su familia. Como parte del tratamiento, se realizó una valoración inicial y se plantearon unos objetivos específicos de fisioterapia acordes con el momento evolutivo del paciente. Se llevó a cabo el tratamiento utilizando diferentes técnicas, que se describirán más adelante, y se evaluaron los resultados. Tras realizar dicha evaluación, se encontró que el tratamiento había resultado eficaz pues, a pesar de la existencia de un breve período de regresión, el paciente mantenía las mismas capacidades que conservaba al inicio del tratamiento.
PALABRAS CLAVE:Atrofia espinal, enfermedades neuromusculares, hipotonía, fisioterapia, rehabilitación.
1. INTRODUCCIÓN
La amiotrofia espinal infantil es una enfermedad autosómica recesiva que se caracteriza por la degeneración de las motoneuronas alfa de la médula espinal. Las manifestaciones clínicas se caracterizan por debilidad muscular generalizada, arreflexia osteotendinosa, hipotonía y atrofia muscular proximal simétrica.
El objetivo de este artículo es presentar un caso de amiotrofia espinal infantil, describiendo el tratamiento de fisioterapia realizado y comprobando su eficacia desde la experiencia profesional de la autora como fisioterapeuta en centros escolares de la Junta de Castilla León. Para ello, en primer lugar como marco teórico se describe la literatura científica sobre la amiotrofia espinal infantil. Posteriormente, se presenta el caso clínico mencionado, referenciando tanto el material y método utilizado, como los antecedentes personales y familiares, la evaluación inicial y los objetivos y técnicas de fisioterapia aplicadas. En su última parte, se evalúan los resultados obtenidos y se realizará una discusión acerca de la eficacia de la terapia a modo de conclusión.
2. MARCO TEÓRICO
2.1. Definición
La amiotrofia espinal infantil o atrofia muscular espinal (AME) en la infancia es una enfermedad neuromuscular hereditaria de transmisión autosómica recesiva, caracterizada por la degeneración de las motoneuronas alfa del asta anterior de la médula espinal, con debilidad proximal y simétrica y atrofia progresiva de los grupos musculares(1). Los primeros casos fueron descritos por Werding en 1891 y por Hoffman en 1893(2).
2.2. Clasificación
Teniendo en cuenta la edad de comienzo, los signos clínicos y la gravedad de la enfermedad, esta se ha clasificado en 3 grupos, que se exponen a continuación.
- AME tipo I o enfermedad de Werding-Hoffman. Es la forma más grave. Se inicia antes de los seis meses. Los bebés afectados no llegan a alcanzar la sedestación y suelen fallecer antes de los 2 años.
- AME tipo II o forma intermedia. Comienza entre los 6 y los 18 meses. Se consigue la sedestación y en algún momento llegan a ponerse de pie con ayuda, pero no deambulan. La supervivencia puede sobrepasar los 4 años de vida.
- AME tipo III o enfermedad de Kugelberg-Welander. Es la forma más leve, comienza a manifestarse después de los 18-24 meses. Se adquiere la capacidad de marcha, pudiendo perderse a edades variables. Los pacientes tienen una esperanza de vida cercana a la normalidad.
Debe añadirse la existencia de una forma de aparición tardía, en la segunda o tercera década de la vida, la AME tipo IV. Se caracteriza por debilidad muscular, porque el curso de la enfermedad es mucho más lento y por una esperanza de vida normal(3) (Tabla 1).
Tabla 1. Clasificación de la Amiotrofia Espinal Infantil
|
|
Edad de comienzo |
Evolución |
|
TIPO I (Enfermedad de Werding-Hoffmann) |
0 - 6 meses |
No se sientan |
|
TIPO II (Forma Intermedia) |
6 - 18 meses |
No se ponen de pie |
|
TIPO III (Enfermedad de Kugelberg-Welander) |
> 18 meses |
Se ponen de pie y caminan |
|
TIPO IV (Adultos) |
2ª - 3ª década |
Caminan sin ayuda |
2.3. Epidemiología
La atrofia espinal infantil es la segunda causa de muerte en el grupo de enfermedades autosómicas recesivas después de la fibrosis quística.(4) Tiene una incidencia entre 1/6.000 y 1/10.000 entre los recién nacidos vivos, y es la segunda enfermedad neuromuscular más frecuente en la infancia, después de la distrofia muscular de Duchenne(5).
2.4. Etiología
La etiología de la amiotrofia espinal es genética, monogénica y autosómica recesiva. En muchos casos se describe consanguinidad entre los padres(6). Se produce una alteración en el brazo largo del cromosoma 5, locus D5s39, región q11.2-q13.3. El gen responsable se identificó en 1995 y se lo denominó Survival Motor Neuron (SMN)(7). Dicho gen está duplicado, existiendo una versión telomérica (SMN1) y otra centromérica (SMN2).
La diferencia crítica entre el gen SMN1 y SMN2 es un cambio único de nucleótido en el exón 7, que hace que el gen SMN1 genere el transcrito completo mientras que, en la mayoría de los casos, el transcrito correspondiente al gen SMN2 carezca del exón 7, produciendo de este modo una proteína menos estable(8).Cuando SMN1 está ausente o es disfuncional, genera una disminución de la proteína necesaria para la función y supervivencia de la motoneurona(9) y la cantidad de proteína SMN funcional producida por SMN2 no es suficiente para prevenir la degeneración progresiva de la motoneurona.
El número de copias del gen SMN2 es variable, oscilando entre cero y
cinco. Varios estudios han demostrado una inversa correlación entre el número de copias de SMN2 y la gravedad de la atrofia espinal. En general, los pacientes con AME tipo I tendrían una o dos copias del gen SMN2, mientras que los pacientes con tipo II y III tendrían de 3 a 5 copias(10).
2.5. Diagnóstico
El diagnóstico de sospecha es fundamentalmente clínico. También hay que tener presente los antecedentes familiares y diversos estudios complementarios (Tabla 2). Entre estos estudios está la biopsia muscular, que demuestra la atrofia de las fibras musculares estriadas difusas y en la que se observan fibras aisladas hipertróficas y atrofia muscular neurogénica. La electromiografía muestra fibrilaciones y otros signos de denervación muscular(11), la velocidad de conducción motora se encuentra disminuida por la pérdida de los axones de conducción rápida y las velocidades de conducción sensitiva son normales. El estudio bioquímico permite hallar que la enzima creatinofosfokinasa está elevada en los casos graves. Finalmente, el diagnóstico se confirma con el estudio cromosómico, verificando la ausencia del exón 7 (con o sin deleción del exón 8), ya que en más de un 90% de los casos se presenta una deleción del gen SMN1(12).
Tabla 2. Diagnóstico de la Amiotrofia Espinal Infantil
|
Antecedentes Familiares |
Patrón de herencia autosómico recesivo. |
|
Manifestaciones clínicas |
Hipotonía, arreflexia, atrofia muscular proximal y simétrica, fasciculaciones de los músculos de la lengua. |
|
Biopsia |
Atrofia de fibras musculares estriadas. |
|
Electromiografía |
Fibrilaciones y signos de denervación muscular. Velocidad de conducción motora disminuida. Velocidad de conducción sensitiva normal |
|
Estudio Bioquímico |
Enzima creatinfosfokinasa (CPK) elevada en casos graves |
|
Estudio Cromosómico |
Ausencia del exón 7 del gen SMN1, en el brazo largo del cromosoma 5 en la región q 11.2-q 13.3 |
En este tipo de casos es necesario realizar un diagnóstico diferencial con otras enfermedades congénitas de aparición precoz que cursan con hipotonia(13) y debilidad, como son distrofia muscular congénita, miopatía congénita, miastenia gravis, distrofia miotónica o miopatías metabólicas, entre otras(14).
En la actualidad, para las familias con antecedentes se recomienda realizar un diagnóstico prenatal mediante análisis molecular de ADN fetal en la 9ª ó 10ª semana de gestación para determinar la condición genética del feto.
2.6. Manifestaciones clínicas
La atrofia muscular espinal o amiotrofia espinal infantil clínicamente se caracteriza por hipotonía y debilidad muscular generalizada. La debilidad normalmente es simétrica, más proximal que distal, y mayor en los miembros inferiores. A nivel cognitivo no hay alteraciones. La sensibilidad está conservada. Los reflejos osteotendinosos pueden estar ausentes o disminuidos dependiendo de la gravedad del caso(15).
En la AME tipo I, las manifestaciones clínicas comienzan antes de los seis meses, aunque la enfermedad puede iniciarse antes del nacimiento, cuando en el último trimestre del embarazo las madres notan disminución de los movimientos del feto(16). La hipotonía es severa y en muchos casos no llegan a adquirir el control cefálico. La afectación de las motoneuronas bulbares con frecuencia produce fasciculaciones en la lengua, lo que conlleva a una dificultad para alimentarse(17). Las contracturas pueden estar presentes en el momento del nacimiento, algunas desarrolladas en el útero por disminución de los movimientos fetales. Las alteraciones secundarias suelen ser el desarrollo de escoliosis, la disminución de la capacidad respiratoria y la fatigabilidad.El diafragma generalmente no sufre alteraciones en relación con la musculatura intercostal y abdominal, provocando una respiración paradójica. Muchos niños requieren intubación y traqueotomía debido a su dificultad respiratoria. La muerte suele ser secundaria a una neumonía u otras complicaciones respiratorias(18).
En la AME tipo II, los niños son capaces de adquirir la sedestación, pero no son capaces de caminar independientemente. Los reflejos osteotendinosos están ausentes y es común que presenten temblor fino en las manos. La debilidad axial y proximal provoca incapacidad para mantener el tronco alineado y esto conlleva la aparición de posturas anormales, provocando a la larga la aparición de deformidades como escoliosis y cifosis(19). La debilidad también afecta a los músculos respiratorios, provocando alteraciones que en los casos más graves pueden llegar a necesitar ventilación mecánica(20). Hay una gran variabilidad de casos según la gravedad, desde chicos más débiles que son incapaces de sentarse, tienen problemas respiratorios y escoliosis temprana, a otros más fuertes que tienen la musculatura del tronco y los músculos respiratorios más resistentes.
En la AME tipo III, la debilidad muscular proximal aparece durante la infancia. Los afectados adquieren las habilidades motrices gruesas, aunque suelen tener dificultades para levantarse del suelo, subir escaleras o correr. Generalmente, la deambulación se mantiene, aunque en algunos casos en los que la debilidad es mayor a largo plazo será necesaria la silla de ruedas para desplazarse. Los pacientes que pierden la deambulación con frecuencia desarrollan escoliosis y otros problemas médicos relacionados con la movilidad reducida, como obesidad y osteoporosis(21).
2.7. Tratamiento
2.7.1. Farmacológico
No existe un tratamiento que remedie la amiotrofia espinal infantil. Sin embargo, existen algunos medicamentos o compuestos farmacéuticos que, a pesar de no curar la enfermedad, pueden aliviar parte de las manifestaciones clínicas. Se han realizado estudios evaluando la eficacia de los mismos en el curso de la enfermedad, teniendo como principio la protección de las neuronas motoras del asta anterior de la médula espinal y la potenciación de SMN2. Medicamentos como Riluzole y Gabapentina, que actúan inhibiendo receptores del glutamato (neurotransmisor excitatorio que produce la muerte neuronal); Creatina , para mejorar el metabolismo energético; Carnitina, por su efecto neuroprotector; y Albuterol , por sus propiedades anabólicas y el efecto molecular sobre la expresión del gen SMN2(22). El grupo de compuestos Inhibidores de la Histona Deacetilasa (HDAC) pueden aumentar la producción del SMN2 así como de las proteínas, histonas y factores de transcripción. Otros medicamentos con este efecto son la Hidroxiurea y el Ácido Valproico (23). En la actualidad, se están llevando a cabo ensayos clínicos para evaluar la eficacia de estos compuestos.
2.7.2. Fisioterápico
El tratamiento de fisioterapia está encaminado a tratar las alteraciones secundarias y complicaciones que aparecen en la evolución de la enfermedad. Estas repercuten a nivel funcional y algunas como las escoliosis o los trastornos respiratorios influyen directamente en el pronóstico vital.(24) Los objetivos del tratamiento fisioterápico, teniendo en cuenta las diferentes formas de la enfermedad y de las complicaciones que conlleva, de forma general se centran en los expuestos a continuación.
- Cuidados y fisioterapia respiratoria. Se debe hacer un programa preventivo para mantener en buenas condiciones la musculatura implicada en la respiración y evitar posibles complicaciones cuando el paciente sufra un problema respiratorio. Se aplicaran técnicas de fisioterapia respiratoria y se tratarán adecuadamente las infecciones agudas del tracto superior. En las fases más avanzadas se aplicará ventilación mecánica no invasiva (NIV) con mascarilla nasal. Los casos más graves requieren intubación(25).
- Control postural. Con el objetivo de que los pacientes puedan mantener un correcto alineamiento de cabeza, tronco y para evitar la fatiga muscular habrá que recurrir a las adaptaciones posturales destinadas a optimizar una sedestación alineada, combinándolas con diferentes cambios posturales como el uso de planos laterales o el uso de hamacas.
- Mantener la fuerza muscular y las actividades motrices que el paciente conserve. Las sesiones deben ser cortas para evitar la fatiga.
- Prevenir la aparición de contracturas, sobre todo en miembros inferiores en aquellos niños que pasen mucho tiempo en sedestación.
- Mantenimiento de la flexibilidad. Mediante movilizaciones pasivas y estiramientos, y en especial para evitar contracturas en la cadera, rodillas, codos y pies.
- Prevenir actitudes posturales incorrectas, para evitar, en lo posible la aparición de deformidades, como la escoliosis. Valorar como interfieren las posturas anómalas en la sedestación funcional del paciente y proporcionar adaptaciones para mantener la columna en posturas correctas. Las escoliosis en las enfermedades neuromusculares generalmente son curvas dorso-lumbares y son más severas en niños que no deambulan, aparecen a edades tempranas y tienen una rápida progresión después de la pubertad.(21) El tratamiento de elección es quirúrgico(26), suele indicarse corsé o asiento moldeado con el objetivo de ayudar a una correcta estabilización del tronco en sedestación y retrasar la cirugía. Se consigue mejorar la función cardiopulmonar, el equilibrio en sedestación, la apariencia y la calidad de vida.
- Uso de adaptaciones posturales para disminuir el efecto de la gravedad en la deformidad, sobre todo en sedestación y bipedestación.
- Programas de bipedestación para evitar a largo plazo fracturas en extremidades inferiores consecutivas a la falta de carga en bipedestación. Hay menor incidencia de displasias de cadera cuando el niño realiza programas de bipedestación con un correcto alineamiento. Se puede utilizar un standing , férulas de yeso con apoyo posterior o bitutores largos con apoyo isquiático.
- Fomentar el mayor grado de independencia del niño, tanto en habilidades manipulativas como en las de movilidad. Suelen ser candidatos a usar silla de ruedas eléctrica.
- Asesoramiento sobre ayudas técnicas y sobre modificación del entorno: supresión de barreras arquitectónicas, uso del ordenador, etc.(27)
- Asesoramiento sobre actividades recreativas terapéuticas, como la actividad acuática, que es terapéutica y lúdica y complementa el tratamiento fisioterápico(28).
2.8 Escalas de Valoración
Las escalas de valoración son un instrumento eficaz para evaluar la función en enfermedades neuromusculares. En la actualidad, se utilizan principalmente para la valoración de resultados en los ensayos clínicos(29). Existen diferentes escalas. Para los pacientes que deambulan, destacan la Minute Walked Test , Timed step o Step Activity Monitor . En niños que no deambulan se utilizan escalas de valoración funcional. Hay dos tipos: las genéricas, para todo tipo de enfermedades neuromusculares, como son la de Vignos , Brooke o la Hammersmith Motor Ability Score (HFMS); y las específicas, que se aplican exclusivamente a una enfermedad determinada. Recientemente se han desarrollado nuevas escalas, como la Motor Function Measure, Egen Clasification Scale o la North Star y diferentes versiones de la Hammersmith Motor Ability Score (HFMS), de la que actualmente se ha hecho una validación de la versión española, específica para la a amiotrofia espinal infantil(30).
3. MATERIAL Y MÉTODO
Para la realización de la búsqueda bibliográfica sobre la amiotrofia espinal infantil se ha hecho una búsqueda en las páginas web de las bases de datos biomédicas MEDLINE PUBMED, PEDro, Cochrane library, IME (Índice Médico Español) y Wolters kluwer/Ovid SP. Y en las bases de datos de la Universidad de Salamanca (USAL), de la Universidad de Cantabria (UNICAN) y de la biblioteca Marquesa de Pelayo (Correspondiente al Hospital Universitario Marqués de Valdecilla, dependiente del Servicio Cántabro de Salud). En dicha búsqueda, se utilizaron como palabras clave: atrofia espinal/spinal atrophy, enfermedades neuromusculares/neuromuscular diseases, hipotonía/hypotonia, rehabilitación/rehabilitation y fisioterapia/physiotherapy.
La documentación primaria del caso clínico está basada en los informes de especialistas del Hospital “General de Segovia”, perteneciente al SACYL (Sanidad de Castilla y León), de los Hospitales “La Paz” y “Ramón y Cajal” pertenecientes al SERMAS (Servicio Madrileño de Salud), y en los informes y valoraciones de fisioterapia realizadas en el colegio al que asistía el paciente objeto de estudio. Deben añadirse, además, las radiografías y el material fotográfico tomados durante el estudio del caso.
El tiempo de este estudio abarca desde septiembre de 2011 a mayo de 2012, periodo en que el paciente tenía 13 años. Sin embargo, el paciente recibía tratamiento de fisioterapia desde los dos años de edad, fecha en que le fue diagnosticada la enfermedad.
3.1. CASO CLÍNICO
El presente caso clínico muestra la historia de un paciente de 13 años y 2 meses de edad, diagnosticado de amiotrofia espinal infantil a los dos años de edad. Se encuentra escolarizado en el colegio de su localidad, que es el lugar donde se ha llevado a cabo el tratamiento fisioterapéutico. El colegio cuenta con 72 alumnos y está localizado en una pequeña población de 929 habitantes.
3.1.2. Antecedentes personales
Madre sana, embarazo normal, parto eutócico, a término, sin problemas. Peso: 3,500g. Talla: 50cm. PC: 34 cm. Apgar 10/10. Movimientos fetales normales. Cuando tiene 21 meses de edad acuden a consulta de pediatría por retraso en la deambulación y dificultades de la marcha, que es balanceante, se incorpora con ayuda, incluso parece haber regresión, tiene dificultad para subir escalones. Desde pediatría, ante la sospecha de una enfermedad neuromuscular, es derivado al servicio de neurología, donde, con 23 meses de edad se le realiza la siguiente valoración:
Atento. Vigil, “muy irritable”. Normoconfigurado. Psiquismo normal. Tono muscular: Hipotonía de miembros inferiores de predominio proximal, muy discreta disminución en miembros superiores. No limitaciones articulares, sedestación estable con buen control de tronco. Moviliza las cuatro extremidades simétricamente. ROT abolidos (patelar, Aquileo, biccipital y triccipital). RCP indeterminado. Flacidez muscular de predominio en cinturas. Marcha con ayuda con balanceo pélvico, con hiperlordosis lumbar. Paso de bipedestación a sedestación con apoyo y muy dificultoso. Gowers (+). Fondo de ojo: papilas y máculas normales. Sensibilidad aparentemente normal. Temblor fino de intención. Se le realizan las siguientes exploraciones complementarias:
- EMG: Trastorno denervativo crónico en músculos explorados.
- VCM y VCS: Normales en nervios distales.
- Hemograma: Normal.
- Bioquímica: Normal, salvo CPK: 230, aldolasa: 7,2.
- Estudio molecular SMN: El paciente porta la delección homocigotadel gen SMN. El estudio de marcadores polimórficos de padres permite realizar la de los otros miembros de la familia.
Se confirmó el diagnóstico de amiotrofia espinal infantil, en este caso es límite entre los tipos II y III.
3.1.3. Antecedentes familiares
Padres no consanguíneos en varias generaciones. Abuelos paternos y maternos viven sin patología. Dos hermanos sanos. Una vez que fue confirmado el diagnóstico, se realizó un estudio genético a la familia. En el mismo se puede ver que los individuos portadores, estos tienen un riesgo aproximado de tener un hijo con AME de 1/300.
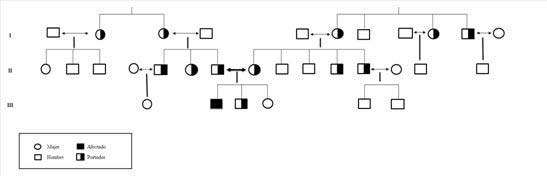
Figura 1.Árbol genealógico del paciente . Es posible observar que es el único afectado de amiotrofia espinal infantil en la familia. En el esquema se representan los individuos portadores. El paciente es el mayor de tres hermanos, a los dos pequeños les hicieron el diagnóstico prenatal para ver si podrían desarrollar la enfermedad. El segundo hermano es portador y en la hermana pequeña está por determinar.
3.1.4. Exploración inicial (27/09/11)
- Balance Articular:
En los miembros superiores: Completo.
En los miembros inferiores: Limitado en caderas, rodillas y tobillos. Flexo de rodillas bilateral irreductible de30º y un flexo de cadera de 50º, (prueba de Thomas). En Tobillos: Derecho: Flexión dorsal 70º, Flexión plantar 30º; Izquierdo: Flexión dorsal 90º. Flexión plantar 35º.
- Columna:
Escoliosis dorsal izquierda, lumbar derecha. Hipocifosis dorsal. Inclinación pélvica marcada. Escoliosis toraco-lumbar derecha. Elevación de hombro izquierdo. Cifosis en la transición toraco-lumbar. Dolor a la palpación de espinosas lumbo-sacras. La movilidad de columna vertebral no está limitada y es indolora.
- Balance Muscular:
Debilidad en miembros inferiores (MMII) y miembros superiores (MMSS).
MMSS: Dificultad para realizar movimientos en contra de la gravedad. La debilidad es mayor a nivel de la musculatura de la cintura escapular. A nivel distal, en la musculatura de ambas manos la debilidad es menor, donde es capaz de realizar movimientos aplicando una pequeña resistencia.
MMII: Debilidad sobre todo a nivel proximal, le cuesta hacer movimientos en contra de la gravedad. La flexo-extensión de tobillo la realiza en contra de la gravedad, incluso aplicando una pequeña resistencia.
- Hipotonía generalizada.
- Hiperlaxitud.
- Sedestación estable. No se mantiene en bipedestación, con gran esfuerzo se mantiene de rodillas.
- Se desplaza gateando con dificultad, cada vez menos, utilizando con preferencia la silla de ruedas para sus desplazamientos.
- Temblor en las manos.
- No presenta problemas respiratorios.
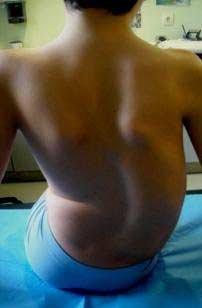
- En las exploraciones complementarias realizadas en el hospital el 26/07/2011 se observó: Tele Rx de columna (26-7-11): Sedestación sin asiento moldeado: Curva D2-D9 izq de 42º .Curva D10-L5 dcha de 65º. Disbalance pélvico: 28 mm, más alta la cresta ilíaca izquierda. Risser 0. Subluxación de ambas caderas. Osteopenia radiológica en rodillas.
- Se realizó un nuevo control radiográfico en noviembre, en el que se observó un importante agravamiento de la curva. (Fig.2)
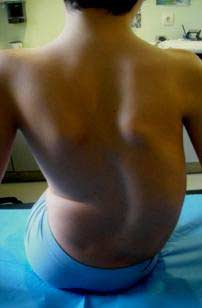
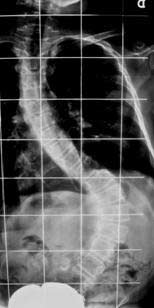
Figura 2 . Escoliosis dorso-lumbar derecha dos meses después de la exploración inicial. Se puede ver el agravamiento de la curva. Escoliosis toraco-lumbar derecha de D10 a L4 de 84º Cobb, con ápex en L1. Escoliosis torácica izquierda de D1 a D10 de 41º. Rotación vertebral de 35º Perdriolle. Inclinación pélvica de 24º, Desequilibrio a la derecha de 16 milímetros. Risser 1.
3.1.5. Objetivos y Tratamiento
Los objetivos que se plantearon estaban dirigidos a disminuir las complicaciones que conlleva la progresión de la enfermedad, adaptados a las características específicas y al momento evolutivo del niño, se centraron en: mantenimiento de la flexibilidad y prevenir la aparición de contracturas; mantener y mejorar la fuerza muscular y las actividades que conserva; corrección postural; evitar la aparición de deformidades y que se agraven las ya existentes; mejorar la función respiratoria; programas de bipedestación; fomentar el gateo y otras formas de desplazamiento; asesoramiento sobre la terapia en el medio acuático: hidroterapia; asesoramiento sobre ayudas técnicas y sobre modificación del entorno; fomentar el mayor grado de independencia del niño; y coordinación con la familia y resto de profesionales del centro.
Se comenzó el tratamiento en septiembre de 2011. El paciente no tomaba ningún tipo de medicación. Se realizaron dos sesiones semanales de fisioterapia y una de hidroterapia. Las sesiones fueron de poca intensidad y duración para evitar llegar a la fatiga. Según los objetivos propuestos el tratamiento que se llevó a cabo fue el siguiente.
- Mantenimiento de la flexibilidad y prevención de la aparición de contracturas, mediante: a) cinesiterapia pasiva y activa-asistida de miembros inferiores (MMII); b) cinesiterapia activa-asistida y activa-resistida de miembros superiores (MMSS); c) estiramientos analíticos de la musculatura de MMII (isquiotibiales, tricéps sural, psoas iliáco, adductores, etc.); y d) ejercicios de flexibilización de columna.
- Mantenimiento y mejora de la fuerza muscular y de las actividades que conservaba mediante: a) ejercicios de tonificación y resistencia de todos los grupos musculares de MMII y de MMSS (MMII, ejercicios activos y activo-asistidos, y MMSS, ejercicios autopasivos, activos y resistidos con diferentes materiales: thera-band , pesos, balones, etc.); y b) ejercicios en cuadrupedia.
- Uso de adaptaciones posturales. Corrección postural mediante el uso de férulas tipo “Rancho del los amigos” para prevenir el equino y con el uso de órtesis para extensión de MMII por la noche. Se realizaron adaptaciones en la silla, en la que lleva un asiento moldeado para evitar que se agraven la oblicuidad pélvica y la escoliosis.
- Evitar la aparición de nuevas deformidades y que se agravasen las ya existentes. Mediante control postural, se realizaron adaptaciones posturales en la silla para evitar el empeoramiento de la oblicuidad pélvica y la escoliosis. También se hicieron ejercicios de flexibilización de columna. Debido a la progresión de la curva,se trató quirúrgicamente, (Fig. 3), de esta forma se evitó que continuara evolucionando y las complicaciones estéticas y respiratorias que conllevaba . Estas escoliosis de más de 45º Cobb aumentan marcadamente durante el periodo de crecimiento rápido, y esta era la situación del paciente, con inmadurez ósea y un gran potencial de crecimiento. Sin embargo, cuando termine el crecimiento óseo, esta escoliosis continuará progresando toda la vida, aproximadamente 1,2º al año, lo que provocará una deformidad estética marcada y la aparición de restricción respiratoria.
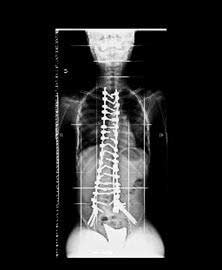
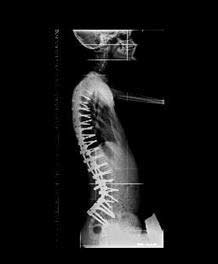
Figura 3. Radiografías de columna tras la cirugía . Se realizó fusión de D2 a iliacos con tornillos iliacos y pediculares en cada nivel . Visiones anteroposterior y lateral.
- Mejora de la función respiratoria. Esto se hizo actuando de forma preventiva, puesto que no presentaba alteraciones. Durante el preoperatorio y postoperatorio a la cirugía de columna se hizo más hincapié en la fisioterapia respiratoria. Algunas de las técnicas utilizadas fueron las siguientes: a) ventilación dirigida; b) ejercicios de expansión costal; c) respiración abdomino-diafragmática; d) espirometria incentivada; y e) ELTGOL (Espiración Lenta Total con Glotis Abierta).
- Programas de bipedestación. Se utilizó un bipedestador o standing en su domicilio, de esta forma se trató de mejorar la cobertura del cotilo de las caderas.
- Fomento del gateo y otras formas de desplazamiento. Se realizaron ejercicios de reptación, en decúbito prono, en cuadrupedia o de rodillas.
- Hidroterapia. En el agua se elimina el efecto de la gravedad y los pacientes no solo son capaces de flotar, sino que pueden hacer una gran cantidad de movimientos. Además, pensamos que los efectos psicológicos subjetivos son importantes. Los pacientes sienten libertad para moverse, la oportunidad de dar rienda suelta a las emociones y el sentimiento de éxito y logro. (Fig. 4)

Figura 4. Ejercicios en piscina.
- Asesoramiento sobre ayudas técnicas y sobre modificación del entorno. El colegio se ha ido adaptando según las necesidades, eliminando las barreras arquitectónicas. Se colocaron rampas en las entradas, un elevador en la escalera para que puediera subir al primer piso (Fig. 5) y en el baño barras de sujeción laterales.

Figura 5. Elevador instalado en la escalera.
- Fomentar el mayor grado de independencia del paciente. Este utiliza silla de ruedas eléctrica para los desplazamientos desde los 9 años, lo cual le ha permitido ser más independiente.
- Finalmente, se trató de coordinar todas estas actividades con la familia y resto de profesionales que se ocupan de la educación del niño, ya que se trata de una labor multidisciplinar.
3.1.6. Resultados
A partir del tratamiento realizado se han obtenido resultados satisfactorios en cuanto a que el paciente no ha perdido de las capacidades que tenia al inicio del estudio. En primer lugar, un mantenimiento del balance articular. Continúa la retracción de rodillas y de cadera y conserva las mismas amplitudes articulares en ambos tobillos. Ha habido incluso un período de regresión, con una disminución del recorrido articular en estas articulaciones debido a la inmovilidad ocasionada tras el postoperatorio de la cirugía de columna. La debilidad ha aumentado de forma general durante ese mismo período, siendo más notable a nivel de MMSS. Posteriormente se ha conseguido recuperar la fuerza que tenía antes de la operación. La columna está más alineada. La función respiratoria se mantiene en buen estado. Los desplazamientos son costosos, gatea con más dificultad, ya le supone un gran esfuerzo. En el resto de síntomas y signos clínicos, hallados en la valoración al inicio del estudio del caso, no hay cambios significativos.
4. CONCLUSIONES
A modo de conclusión general es posible afirmar que el tratamiento de fisioterapia durante este período ha sido efectivo, puesto que el paciente mantiene las capacidades que tenia al iniciarse el mismo. En cuanto al balance articular y muscular ha habido un período de regresión ocasionado por la inmovilización secundaria a la cirugía, recuperando las amplitudes articulares y la fuerza anteriores una vez superada esta fase. La función respiratoria se mantiene en buen estado. Los desplazamientos los realiza con más dificultad. En el resto signos y síntomas que presentaba el paciente no hay cambios significativos.
La ausencia de curación en estas enfermedades no significa que no puedan ni deban ser tratadas. La intervención fisioterapéutica puede prevenir las complicaciones, mantener la función y mejorar la calidad de vida del paciente. Para ello, es importante que exista un diagnóstico precoz y un trabajo en equipo, ya que las enfermedades neuromusculares siguen un patrón predecible de progresión o regresión y las diferencias individuales en el curso de la enfermedad pueden ser significativas.
La intervención sanitaria aunque persigue en primer lugar la sanación del enfermo, también debe preocuparse por la calidad de vida del paciente cuando esto no es posible. La fisioterapia, además de ser una disciplina con una fuerte fundamentación científica, posee una fuerte vocación humanista. Y es precisamente en casos como el tratado donde lo demuestra, tratando de aumentar la calidad de vida de las personas afectadas por enfermedades incurables. Creo que en este caso se muestran las limitaciones, pero también las potencialidades de la fisioterapia como instrumento para ayudar a la mejora en las condiciones de vida de las personas.
BIBLIOGRAFÍA
1. Dubowitz V. Muscle disorders in childhood. London; Saunders: 1978: p.146-190.
2. Rueda SCM, Lancheros EAG, Hernández CR. Síndrome de Werdnig-Hoffmann (atrofia muscular espinal de la infancia). Presentación de un caso y revisión en la literatura . MedUNAB. 2010; 13(2):116-122.
3. Pearn J. Classification of spinal muscular atrophies. Lancet. 1980; 1(8174): 919-922
4. Prior TW, Snyder PJ, Pyatt RE, Mihal DC, Conlan T, Rink BD et al. Newborn and carrier screening for spinal muscular atrophy. Am J Med Genet, Part A. 2010; 152(7):1608-1616.
5. Shawky RM, El-Sayed NS. Clinico-epidemiologic characteristics of spinal muscular atrophy among Egyptians. Egypt J Med Hum Genet. 2011; 12(1): 25-30.
6. Collado-Ortiz MA, Shkurovich-Bialik P, González-De Leo S, Arch-Tirado E. Atrofia espinal tipo I (síndrome de Werdnig-Hoffmann). Reporte de un caso. Cir ciruj. 2007; 75 (2):119-122.
7. Lefebvre S, Burglen L, Reboullet S, Clemeront O, Burlet, P, Viollet L et al.. 1995. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995; 80: 155-165.
8. Tizzano E, Baiget M. Bases moleculares de la atrofia muscular espinal: el gen SMN. Neurología. Barcelona, Spain. 2000; 15(9): 393-400.
9. Prior TW. Perspectives and diagnostic considerations in spinal muscular atrophy. Genetics. 2010;12(3):145-152.
10. Swooboda KJ, Prior TW, Scott CB, McNaught TP, Wrinde MC, Reyna SP et al. Natural history of denervation in SMA: relation to age, SMN2 copy number, and function. Ann Neurol. 2005; 57: 704-712.
11. Han JJ, McDonald CM. Diagnosis and Clinical Management of Spinal Muscular Atrophy. Phys Med Rehabil Clin N Am. 2008; 19(3):661-680.
12. Garófalo Gómez N, Zaldívar Vaillant T, Vargas Díaz J, Rojas Massipe E, Novoa López L. Atrofia muscular espinal en el niño. Rev Cubana Pediatr. 2009; 81(3):0-0.
13. Prats-Viñas J. Enfoque diagnóstico del niño hipotónico. Protocolos Diagnóstico Terapeúticos de la Asociación Española de Pediatría: Neurología Pediátrica. 2008: 71-78. Disponible en: www.aeped.es/protocolos/
14. Medina JS, Moreno González AM, Romero Baizabal BL, Aguilar JH, Delgado CG, Avelar A. Lactante con atrofia muscular espinal y encefalopatía hipóxico-isquémica. Bol Med Hosp Infant Mex. 2010; 67(1): 63-73.
15. Picó G. Atrofia espinal Infantil. Asociación Española de pediatría. Protocolos de diagnóstico terapeúticos: neurología pediátrica. 2008: 79-82. Disponible en: www.aeped.es/protocolos/
16. González De Dios J, Martínez Frías ML, Arroyo Carrera I, Fondevilla Saucí J, Sanchís Calvo A, Hernández Ramón F, et al. Importancia diagnostica de los signos de hipocinesia fetal en la atrofia muscular espinal de presentacion neonatal. An Esp Pediatr. 2002; 56: 233-240.
17. DiVito D, Konek S. Spinal Muscular Atrophy— Summary for Nutritional Care. ICAN: Infant Child Adolesc Nutr. 2010; 2(6):348-354.
18. Schroth MK. Special Considerations in the Respiratory Management of Spinal Muscular Atrophy. Pediatrics. 2009; 123(4): 245-249.
19. Castillo MAC, Gay MP, Ramos MAB, Alvarez AG. Escoliosis en atrofia muscular espinal. Med Rehabil. 2002; 15: 16-18.
20. Ching SY, Wong YQ, Hui JH, Wong HK, Ong HT, T DYG, H. Pulmonary function and scoliosis in children with spinal muscular atrophy types II and III. J Paediatr Child Health. 2003; 39 (9): 673-676.
21. D'Amico A, Mercuri E, Tiziano FD, Bertini E. Spinal muscular atrophy. Orphanet J Rare Dis. 2011; 6(1): 71-81
22. Tizzano F. E. Atrofia muscular espinal: contribuciones para el conocimiento, prevención y tratamiento de la enfermedad y para la organización de familias. Madrid: Real Patronato sobre Discapacidad; 2007.
23. Darbar IA, Plaggert PG, Resende MBD, Zanoteli E, Reed UC. Evaluation of muscle strength and motor abilities in children with type II and III spinal muscle atrophy treated with valproic acid. BMC Neurology. 2011; 11(1): 36-40.
24. Febrer A, Meléndez M. Atrofia muscular espinal. Complicaciones y Rehabilitación. Rehabilitación. 2001; 35 (5): 307-311
25. Sharma GD. Pulmonary Function Testing in Neuromuscular Disorders. Pediatrics. 2009; 123 (Supl 4): 219-221.
26. Mullender MG, Blom NA, De Kleuver M, Fock JM, Hitters WMGC, Horemans AMC, et al. A Dutch guideline for the treatment of scoliosis in neuromuscular disorders. Scoliosis. 2008; 3: 1-14.
27. Fagoaga MJ. Enfermedades neuromusculares. En: Fagoaga MJ., Macias ML. Fisioterapia en Pediatría. 1ª Ed. Aravaca, Madrid: McGraw-Hill Interamericana; 2002. p. 289-297.
28. Cunha MC, Oliveira AS, Labronici RH, Gabbai AA. Atrofia muscular espinhal tipo II (intermediária) e III (Kugelberg-Welander): evoluçao de 50 pacientes com fisioterapia e hidroterapia em piscine. São Paulo. Arq Neuro-Psiquiatr. 1996; 54(3): 402-406.
29. Finkel RS, Hynan LS, Glanzman AM, PT, DPT, PCS, ATP, Owens H, MSPT, Nelson L, MPT, Cone SR, MSPT, et al. The Test of Infant Motor Performance: Reliability in Spinal Muscular Atrophy Type I. Pediatr Phys Ther. 2008; 20(3): 242-246.
30. Febrer A, Vigo M, Fagoaga J, Medina-Cantillo J, Rodríguez N, Tizzano E. Escala de valoración funcional de Hammersmith para niños con atrofia muscular espinal. Validación de la versión española. Rev Neurol. 2011; 53 (11):657-663.